



Data Visualization Fundamentals For Biologists
A Beginner's Guide to Basic Data Visualization in Bioinformatics with Python


An ask: If you liked this piece, I’d be grateful if you’d consider tapping the “heart” 💙 in the header above or sharing this newsletter with the button below. It helps me understand which pieces you like most and supports this newsletter’s growth. Thank you!
🧬 Data Visualization Fundamentals For Biologists
As the readership of the Decoding Biology newsletter has expanded from dozens to thousands of readers, I've engaged in conversations with an increasing number of students and researchers who are interested in learning bioinformatics but have found learning to code daunting. My last newsletter, titled Python Fundamentals For Biologists, aimed to lower the activation energy for those getting started by highlighting the key programming concepts that will appear time and time again.
Today's newsletter is a compliment to Python Fundamentals For Biologists. In this newsletter, I'll provide an introduction to data visualization for bioinformatics without overwhelming beginners with minutiae or glossing over critical concepts. In bioinformatics, data visualization is crucial for interpreting complex biological datasets, making it easier for researchers to identify patterns, trends, and insights. Additionally, effective visualization enhances data-driven decision-making, aids in hypothesis generation, and facilitates communication of findings. Let's begin!
◈◈◈
🧬 Introduction To Matplotlib and Seaborn
Matplotlib and Seaborn are two of the most commonly used libraries for data visualization in Python. Matplotlib provides a MATLAB-like interface and is widely used to create publication-quality visualizations of varying types. Seaborn is built atop Matplotlib and is specialized for statistical data visualization. Additionally, Seaborn provides more aesthetically pleasing defaults compared to Matplot lib. Before diving into specific data visualization techniques, you can install Matploblib and Seaborn in your Python IDE using the following code:
import matplotlib.pyplot as plt
import seaborn as sns◈◈◈
🧬 Identifying Patterns In Data With Scatter Plots
A scatter plot is a type of data visualization that displays individual data points on a two-dimensional (x-y coordinate) or three-dimensional (x-y-z coordinate) graph where each point on the plot represents the values of all variables. Scatter plots make it easy to examine the relationship between variables, and as a result, they are useful for identifying patterns, correlations, and outliers in data. Importantly, scatter plots are only appropriate when all variables are numerical. The code below demonstrates the general syntax for producing scatter plots with Seaborn:
sns.scatterplot(x='Column 1', y='Column 2', data=dataset_name)
plt.xlabel('Column 1')
plt.ylabel('Column 2')
plt.title('Scatter Plot')
plt.show()Note: In the first line of code 'Column 1' and 'Column 2' must match the actual column names in your dataset. Additionally, dataset_name should be replaced with the variable name you used to store your CSV file. The code sample below demonstrates a real-world example of what this may look like:
cancer_by_state = read_csv('actual file path...')
sns.scatterplot(x='Skin Cancer', y='UV Index', data=cancer_by_state)
plt.xlabel('Rate of skin cancer per 1000 people by State')
plt.ylabel('Average UV index by state')
plt.title('Relationship b/w Skin Cancer and UV Index')
plt.show()In bioinformatics, scatter plots are commonly used to visualize the relationship between biological variables. For instance, scatter plots may be used to examine the relationship between gene expression levels before and after a specific intervention. For example, the image below is a figure reproduced from a study titled, Role and mechanism of the AMPK pathway in waterborne Zn exposure influencing the hepatic energy metabolism of Synechogobius hasta:1
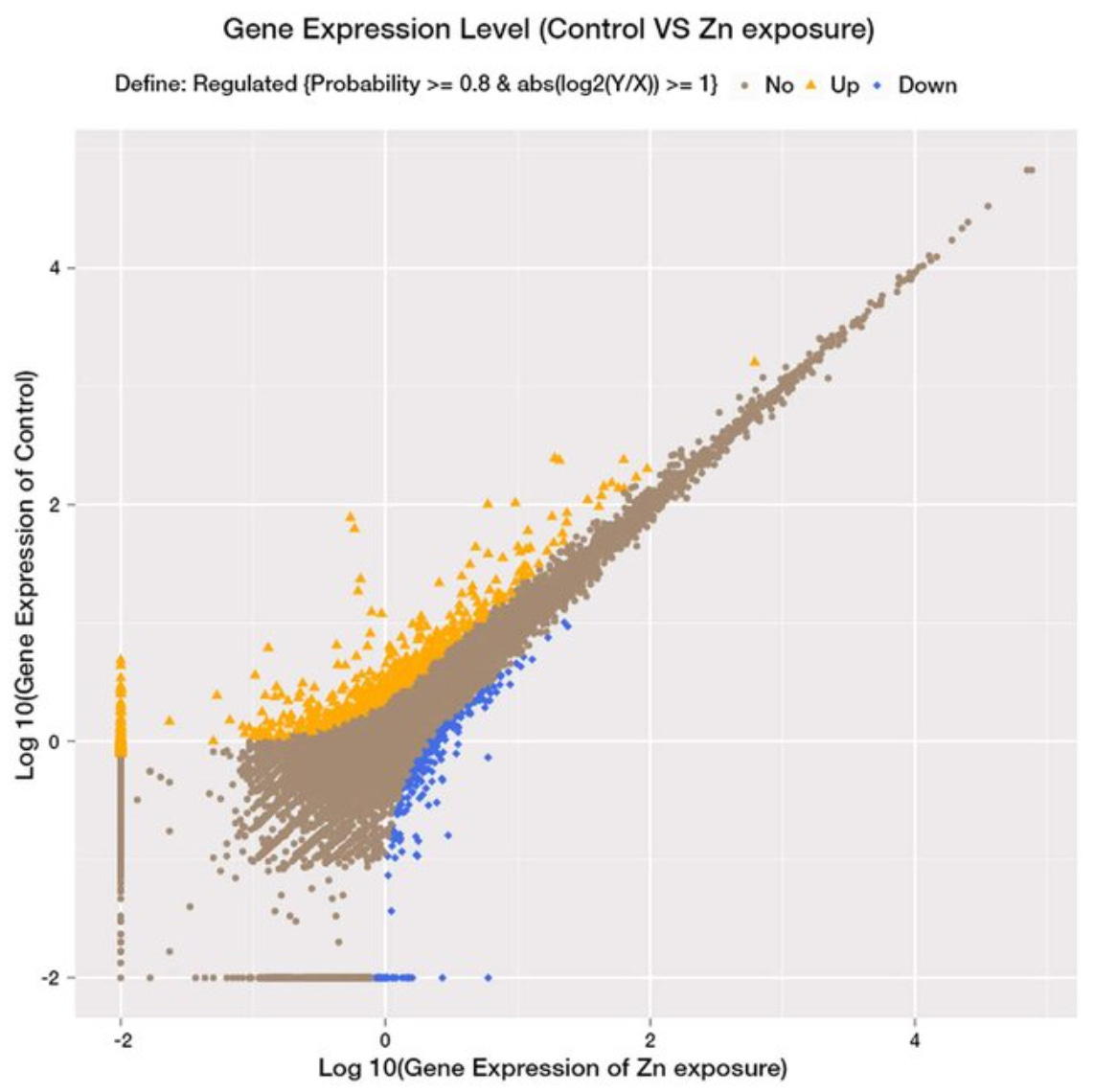
The scatterplot above shows the correlation of gene expression profiles between a control group (y-axis) and a waterborne zinc-exposed (x-axis) group of Synechogobius hasta, a small edible fish species. Genes that are up-regulated in the control group as compared to the zinc-exposed group are indicated in orange, whereas genes that are down-regulated in the control group as compared to the zinc-exposed group are indicated in blue. The genes indicated in brown weren't differentially expressed between the two groups.
◈◈◈
🧬 Visualizing Time Series Data With Line Charts
A line plot is a type of data visualization that displays data points connected by straight-line segments. Line plots are essentially scatter plots where the points are ordered and connected. However, whereas scatter plots are used to understand the relationship between two independent variables, line plots are appropriate when the y-variable is a continuous function of the x-variable. As a result, line plots are commonly used to represent the change in a variable over time. The code below demonstrates the general syntax for producing line plots with Matplotlib:
plt.plot('x_values', 'y_values', data=data=dataset_name)
plt.xlabel('X-axis Label')
plt.ylabel('Y-axis Label')
plt.title('Line Plot Title')
plt.show()Note: In the first line of code 'x_values' and 'y_values ' must match the actual column names in your dataset. Additionally, dataset_name should be replaced with the variable name you used to store your CSV file. The code below provides a real-world example:
muscle_oxygen = read_csv('actual file path...')
plt.plot('Time', 'SmO2', data=muscle_oxygen, color= 'Green')
plt.xlabel('Time (Seconds)')
plt.ylabel('Muscle Oxygenation (%)')
plt.title('Muscle Oxygenation During Exericse')
plt.show()Which produces the following figure:
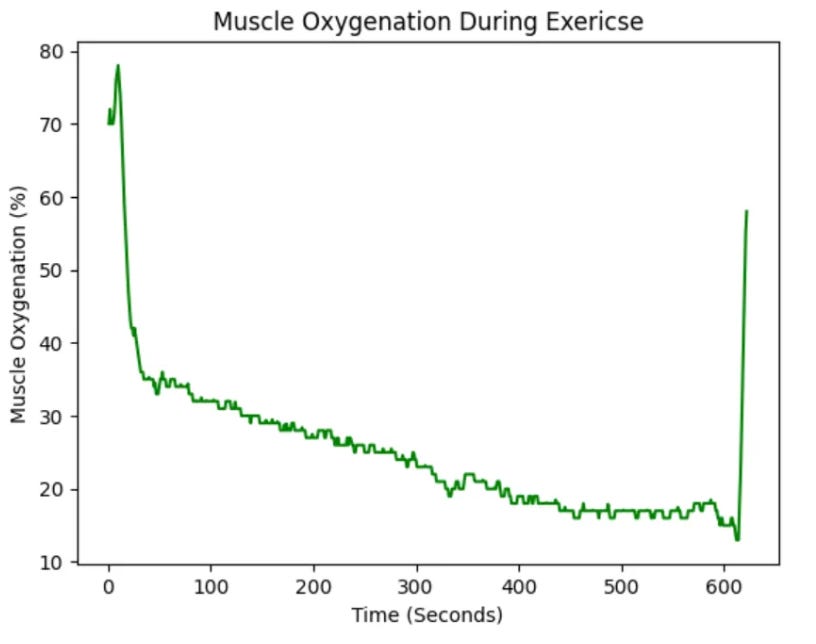
The image above shows muscle oxygenation data recorded with a NNOXX wearable device plotted over time. However, in bioinformatics, line plots are commonly utilized to illustrate trends in gene expression levels over time or under different experimental conditions. This type of plot allows researchers to observe how the expression of a gene evolves across various biological states, as demonstrated in the image below, which appears in a study titled, Gene Expression Trees In Lymphoid Development:2
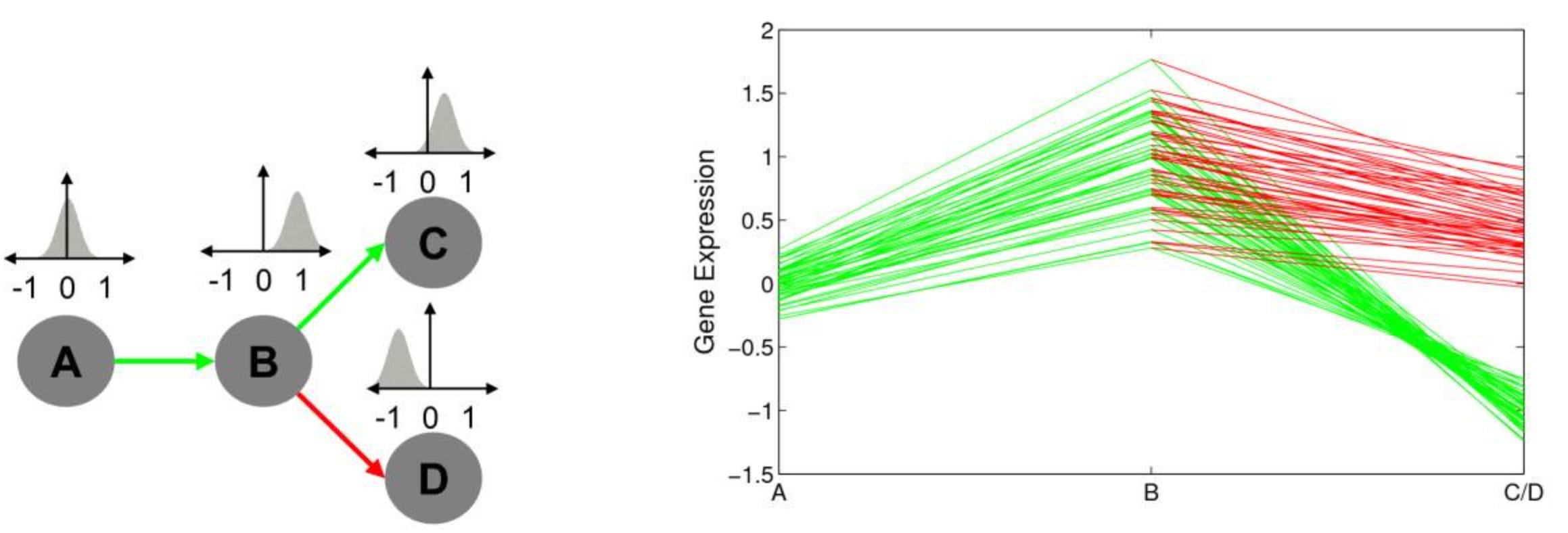
The image above represents the expression pattern of a specific gene across different time points. On the left side of the image, we have a simple development tree, where arrows represent the dependencies between variables. Then, on the right side of the image, we can see gene expression values represented on the y-axis for distinct developmental stages on the x-axis. Additionally, each line on the graph corresponds to the developmental profile of a given gene following one of two developmental paths depicted on the tree (A->B->C or A->B->D). The resulting line graph provides a clear visual depiction of how the gene expression changes throughout development and by superimposing the lines that correspond to path B->C (green) and B->D )reg) we can easily contrast the differences in expression values of genes in these two alternate differentiation pathways.
◈◈◈
🧬 Comparing Categorical Data With Bar Charts
Bar charts are graphical representations used to show the relationship between categorical and numeric variables. The length of each bar on a bar chart is proportional to the value it represents, making it effective for comparing the magnitudes of different categories. The code below demonstrates the general syntax for producing bar charts with Seaborn:
sns.barplot(x='Column 1', y='Column 2', data=dataset_name)
plt.xlabel('Column 1')
plt.ylabel('Column 2')
plt.title('Scatter Plot')
plt.xticks(rotation=70)
plt.show()Note: In the first line of code 'Column 1' and 'Column 2' must match the actual column names in your dataset. Additionally, dataset_name should be replaced with the variable name you used to store your CSV file. The code sample below demonstrates a real-world example of what this may look like:
Virus_database = read_csv('actual file path...')
sns.barplot(x ='Family', y = 'Average Radius', data = Virus_database)
plt.xlabel('Virus Family')
plt.ylabel('Average Radius')
plt.title('Average Radius Per Virus Family')
plt.xticks(rotation=80)
sns.set(style="darkgrid")#Change background color
plt.show()Which produces the following figure:

In bioinformatics, bar charts are used to compare the abundance or frequency of different biological entities, such as genes, proteins, or mutations, across various conditions or samples or to compare a specific, quantifiable characteristic between different organisms, as demonstrated in the image above. Alternatively, bar charts can represent the mean expression of varying genes under different treatment conditions. In this example, each bar corresponds to a specific gene, and the height of the bar indicates the average expression level as demonstrated in the image below, which appears in a study titled, LRRTM3 interacts with APP and BACE1 and has variants associating with late-onset Alzheimer's disease:3

The image above compares the expression level of six different genes, listed on the x-axis, in the brains of wild-type (WT), Lrrtm3 knock-out (LRRTM3 K/O), and heterozygote (LRRTM3 HET) mice. The three mouse genotypic groups are color-coded, as shown in the upper left corner of the image. Additionally, the vertical displacement of each bar depicts the mean gene expression level for that group, and the error bars represent the standard deviations from the average of all measurements made in all of the mice in each group.
◈◈◈
🧬 Understanding Data Distribution With Histograms
Histograms are graphical representations of how numerical or categorical data are distributed in a dataset. When working with numerical data, data is binned into continuous ranges, and then bars are used to represent the frequency or count of data points within each bin. When working with categorical data, the bins are instead composed of the varying categories in the dataset. The code below demonstrates the general syntax for producing bar histograms with Seaborn:
sns.histplot(dataset_name['column'], bins=20, kde=True)
plt.xlabel('Bin names')
plt.ylabel('Frequency')
plt.title('Histogram')
plt.show()Note: In the first line of code, dataset_name should be replaced with the variable name you used to store your data and 'column' should be replaced with an actual column name in your dataset. If your data is numerical, the code bins = # determines how many bins the data is split into (this code does not need to be included if the data is categorical). Then, the code kde=True computes a kernel density estimate to smooth the distribution and show on the plot as (one or more) line(s). However, this is only relevant to univariate data. The code sample below demonstrates a real-world example of what this may look like:
Virus_database = read_csv('actual file path...')
sns.histplot(Virus_database['Average Radius'], bins=20, kde=True)
plt.xlabel('Average Radius')
plt.ylabel('Frequency')
plt.title('Distriution Average Virus Radius')
plt.xticks(rotation=90)
plt.show()Which produces the following chart:

The image above represents the distribution of the average virus radius in a database containing information about various virus families. You’ll note that the data is positively skewed (right-skewed), which is when the distributions tail is longer on the right side and its peak is on the left side.
In bioinformatics, histograms are often used to visualize the distribution of gene expression levels, protein concentrations, or sequence lengths and provide insights into the underlying patterns and frequencies within a dataset, as demonstrated in the figure below, which is from a study titled, Transcriptome Sequencing Revealed Significant Alteration of Cortical Promoter Usage and Splicing in Schizophrenia:4

The histogram in the image above shows the distribution of gene expression value for genes in representative samples C26 and S26. The gene expression levels were first normalized to RPKM (reads per Kilobase of transcription per million mapped reads) values and then log2 transformed as shown on the x-axis. The y-axis, on the other hand, represents the frequency of genes with expression values within the specific log2 RPKM intervals represented on the x-axis.
◈◈◈
🧬 Boxplots for Outlier Detection
A boxplot, also known as a box-and-whisker plot, is a graphical representation of the distribution of a dataset that summarizes key statistical measures, including the median, quartiles, and potential outliers, as demonstrated in the image below:

Now, the code below demonstrates the general syntax for box and whisker plots with Seaborn:
sns.boxplot(x='category_column', y='numeric_column', data=dataset_name)
plt.xlabel('Category')
plt.ylabel('Numeric Value')
plt.title('Box Plot')
plt.show()Note: In the first line of code 'category_column' and 'numeric_column' must match the actual column names in your dataset. Additionally, dataset_name should be replaced with the variable name you used to store your CSV file. The code sample below demonstrates a real-world example of what this may look like:
dementia_dataset = read_csv('actual file path...')
sns.boxplot(x='CDR', y='Age', data= dementia_dataset)
plt.xlabel('Clinical Dementia Rating')
plt.ylabel('Age')
plt.title('Age Distribution Based On CDR')
plt.xticks(rotation=90)
plt.show()Which produces the following figure:

The figure above shows the distribution of ages for individuals with clinical dementia ratings (CDR) between 0, indicating no dementia, and 2.0, indicating moderate cognitive impairment. Notice that the median age increases from a CDR rating of 0.0 (~71 years) to 2.0 (~82 years). Additionally, you can see that the interquartile range and size of the whiskers decrease as you move from a low to a high CDR rating. In essence, this tells us that there is much more age variability among individuals with a low CDR rating than individuals with a high CDR rating.
In bioinformatics, boxplots may be used to compare the distribution of a quantitative variable across different experimental groups or conditions. For example, imagine a study examining the expression levels of a set of genes under various treatment conditions. A boxplot can be created for each gene to visualize the distribution of expression levels in response to different treatments, allowing researchers to compare the variability in gene expression between conditions, as demonstrated in the figure below from a study titled, Deconstructing transcriptional variations and their effects on immunomodulatory function among human mesenchymal stromal cells:5

In the study referenced above, the research investigated variations in gene expression among mesenchymal stem cells (MSCs) exposed to inflammatory cytokine INFγ to understand how these gene expression changes influence MSCs' ability to regulate or modify the activity of the immune system. The boxplot above shows the baseline (labeled Untreat) expression level for eight genes compared to the level of expression following INFγ exposure (labeled Treat). Notably, the expression level of all genes increased in response to INFγ exposure, but the magnitude of change varied from gene to gene. The box and whisker plot demonstrates the interquartile range, range of data points, and outliers from each gene under each condition.
◈◈◈
🧬 Heat Maps for Correlation
A heatmap presents data in a matrix format, where values are represented as colors, and the intensity of the color indicates the strength of a correlation between two variables. The code block below demonstrates the general syntax for producing a heatmap:
correlation_matrix = dataset_name.corr()
sns.heatmap(correlation_matrix, annot=True, cmap='coolwarm')
plt.title('Correlation Heatmap')
plt.show()Note: In the first line of code 'dataset_name' should be replaced with the variable name you used to store your CSV file. The code sample below demonstrates a real-world example of what this may look like:
dementia_dataset = read_csv('actual file path...')
correlation_matrix = dementia_dataset.corr()
sns.heatmap(correlation_matrix, annot=True, cmap='coolwarm')
plt.title('Oasis-1 Brain Dataset Heatmap')
plt.show()Which produces the following figure:
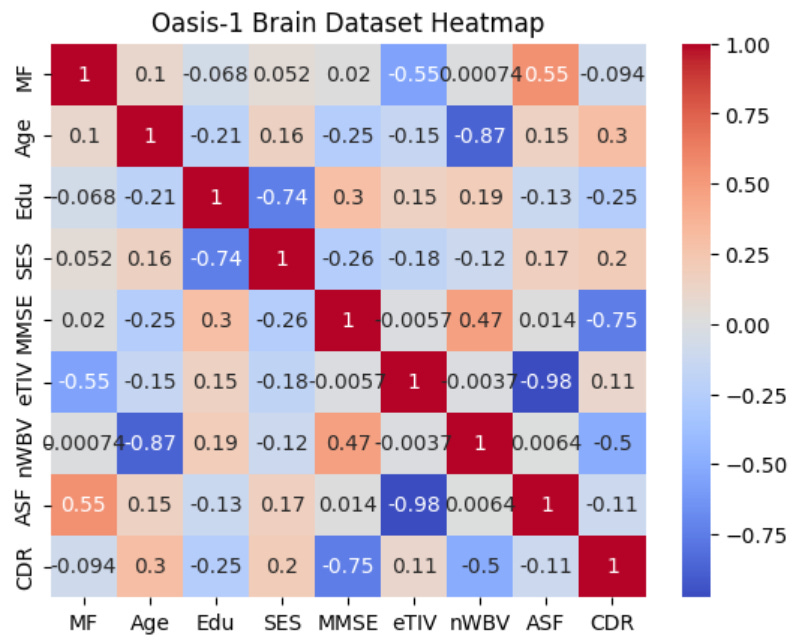
The heat map in the image above shows the correlations between variables in the Oasis-1 brains dataset. The more intensely warm a cell in the matrix is, the stronger the positive association between two variables, and the more intensely cool a cell in the matrix is, the stronger the negative association between two variables. For example, we can see that the cell at the intersection between AFF and eTIV is dark blue, and within the cell, we see a correlation value of -0.98, which is a near-linear inverse correlation.
In bioinformatics, heatmaps are commonly used to represent large-scale datasets, such as gene expression profiles, where rows correspond to genes and columns correspond to samples or experimental conditions. Each cell in the heatmap represents the expression of a specific gene in a specific sample, with colors indicating the expression intensity. This visual representation enables researchers to quickly identify patterns of upregulation, downregulation, or co-regulation across multiple genes and experimental conditions, as demonstrated in the figure below from a paper titled, Maternal Experience with Predation Risk Influences Genome-Wide Embryonic Gene Expression in Threespined Sticklebacks (Gasterosteus aculeatus):6
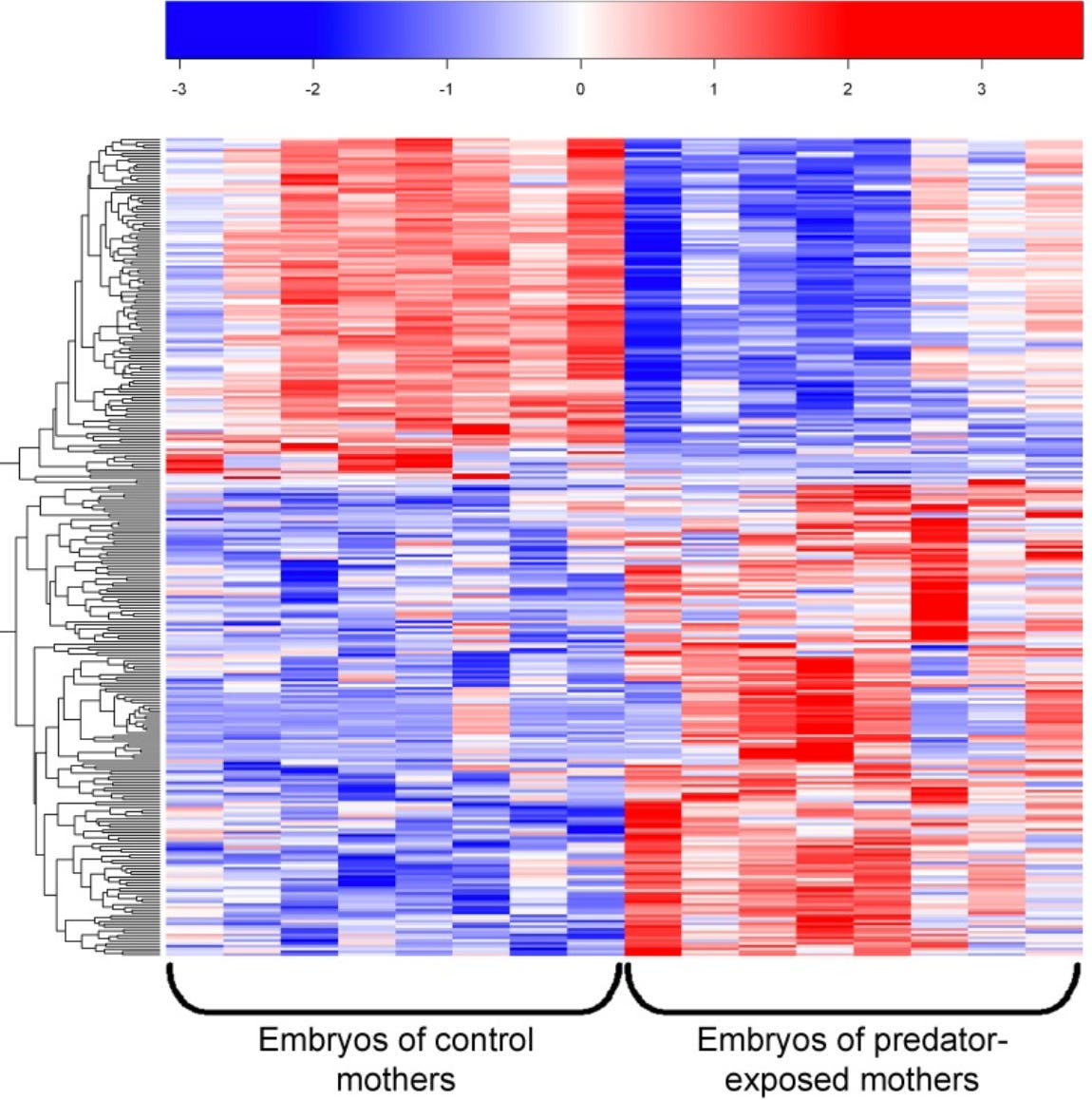
In the study referenced above, researchers sought to investigate the effect of maternal exposure to predation risk on the embryonic transcriptome in stickleback fish. To do this, the researchers used RNA-sequencing to compare genome-wide transcription in three-day post-fertilization embryos of both control and predator-exposed stickleback mothers. The image above depicts a heatmap showing the general pattern of gene regulation for 295 genes that were differentially expressed due to maternal exposure to predation risk. For example, we can see that the genes in the upper left quadrant of the heatmap were up-regulated in the control group but down-regulated in the predator-exposed group (upper right quadrant). In contrast, the genes in the lower left quadrant were down-regulated in the control group but up-regulated in the predator-exposed group (lower right quadrant). Thus, by performing this analysis, biological pathways involved in metabolism, epigenetic inheritance, and neural proliferation and differentiation that differed between treatments were revealed.
◈◈◈
🧬 Customizations and Combining Visualizations
Often, when visualizing complex biological data, you'll want to combine multiple subplots into a single graph so you can view data side-by-side and better understand the story it's telling. The code block below demonstrates the generic syntax for producing subplots within a figure:
plt.subplot(2, 2, 1) #Two plots, 2 columns, first plot
plt.plot(x='column_1', y='column_2', data=dataset_name)
plt.subplot(2, 2, 2) #Two plots, 2 columns, second plot
plt.plot(x2, y2)
plt.show()Note: In the first/third line of code you should list the number of plots and columns that are relevant to your analysis. If you want more than two subplots you’ll need to repeat the plt.subplot( ) code for each newly added plot. Additionally, 'dataset_name' should be replaced with the variable name you used to store your CSV file. The code block below demonstrates a real-world example of what this may look like:
#Load dataset
dementia_dataset = read_csv('actual file path...')
#Arrange sizing and spacing of subplots
plt.subplots(figsize=(12, 8)) #
plt.subplots_adjust(hspace = .2, wspace=.2)
#Create first subplot
plt.subplot(2, 2, 1)
correlation_matrix = dementia_dataset.corr()
sns.heatmap(correlation_matrix, annot=False, cmap='coolwarm')
plt.xlabel('Features')
plt.ylabel('Features')
plt.title('Oasis-1 Brain Dataset Heatmap')
#Create second subplot
plt.subplot(2, 2, 2) # Two plots, one column, second plot
sns.boxplot(x='CDR', y='Age', data= dementia_dataset)
plt.xlabel('Clinical Dementia Rating')
plt.ylabel('Age')
plt.title('Age Distribution Based On CDR')
plt.xticks(rotation=90)
#Show subplots
plt.show()Which, produces the following figure:

◈◈◈
Want To Learn More? Check Out The Following Related Newsletters!



Wu K, Huang C, Shi X, Chen F, Xu YH, Pan YX, Luo Z, Liu X. Role and mechanism of the AMPK pathway in waterborne Zn exposure influencing the hepatic energy metabolism of Synechogobius hasta. Sci Rep. 2016 Dec 9;6:38716. doi: 10.1038/srep38716. PMID: 27934965; PMCID: PMC5146659.
Costa IG, Roepcke S, Schliep A. Gene expression trees in lymphoid development. BMC Immunol. 2007 Oct 9;8:25. doi: 10.1186/1471-2172-8-25. PMID: 17925013; PMCID: PMC2244641.
Lincoln S, Allen M, Cox CL, Walker LP, Malphrus K, Qiu Y, Nguyen T, Rowley C, Kouri N, Crook J, Pankratz VS, Younkin S, Younkin L, Carrasquillo M, Zou F, Abdul-Hay SO, Springer W, Sando SB, Aasly JO, Barcikowska M, Wszolek ZK, Lewis JM, Dickson D, Graff-Radford NR, Petersen RC, Eckman E, Younkin SG, Ertekin-Taner N. LRRTM3 interacts with APP and BACE1 and has variants associating with late-onset Alzheimer's disease (LOAD). PLoS One. 2013 Jun 4;8(6):e64164. doi: 10.1371/journal.pone.0064164. PMID: 23750206; PMCID: PMC3672107.
Wu JQ, Wang X, Beveridge NJ, Tooney PA, Scott RJ, Carr VJ, Cairns MJ. Transcriptome sequencing revealed significant alteration of cortical promoter usage and splicing in schizophrenia. PLoS One. 2012;7(4):e36351. doi: 10.1371/journal.pone.0036351. Epub 2012 Apr 27. PMID: 22558445; PMCID: PMC3338678.
Sun C, Zhang K, Yue J, Meng S, Zhang X. Deconstructing transcriptional variations and their effects on immunomodulatory function among human mesenchymal stromal cells. Stem Cell Res Ther. 2021 Jan 9;12(1):53. doi: 10.1186/s13287-020-02121-8. PMID: 33422149; PMCID: PMC7796611.
Mommer BC, Bell AM. Maternal experience with predation risk influences genome-wide embryonic gene expression in threespined sticklebacks (Gasterosteus aculeatus). PLoS One. 2014 Jun 2;9(6):e98564. doi: 10.1371/journal.pone.0098564. PMID: 24887438; PMCID: PMC4041765.