



Python Fundamentals For Biologists
A Concise Primer for Beginners


Decoding Biology is now read in 49 US states and 91 countries. Thank you to all of my readers for your support! If you enjoy today’s newsletter, please consider tapping the ❤️ in the header above. It helps me understand which pieces you like most and supports this newsletters continued growth. Thank you!
🐍 Python Fundamentals For Biologists 💻
As the Decoding Biology newsletter has grown, I've spoken to more and more students and researchers who find themselves at a crossroads in the rapidly evolving field of bioinformatics, where big data is driving groundbreaking discoveries. While the allure of delving into the world of programming to unlock new insights is strong, the path forward can be daunting, especially for those with limited programming backgrounds.
Today's newsletter is for my readers who are interested in learning bioinformatics but are overwhelmed by the sheer volume of coding instructionals available. This guide aims to highlight key concepts without overwhelming beginners with minutiae or glossing over critical concepts.
Importantly, this guide assumes you are familiar with basic Python data types and syntax. With that foundation, you'll be ready to learn key programming concepts that will appear time and time again. From understanding the logic behind loops and conditionals to deciphering the power of functions, this guide is your short go-to resource, equipping you with the tools to decipher and compose code that unlocks the mysteries hidden in biological data.
◈ ◈ ◈
🐍 Storing Data In Lists and Dictionaries 💻
Lists and dictionaries play pivotal roles in managing and manipulating biological data. In simple terms, a list is like an ordered checklist where each item has a discrete position, and a dictionary is like a set of labeled boxes where each item has a unique label. Lists are good for storing sequences of items, while dictionaries are great for associating information with specific labels or keys. Bioinformatics tasks, such as sequence analysis, rely heavily on these fundamental data structures to navigate and manipulate biological datasets efficiently.
Working With Lists:
A list is a built-in data structure that allows you to store and organize a collection of objects, including integers, strings, and even other lists. In bioinformatics, DNA, RNA, and protein sequences are commonly stored as lists. Lists are defined with square brackets, [ ], and filled with objects separated by commas. Additionally, lists are ordered and mutable, meaning you can change their content by adding or removing elements.
Bioinformatics tasks frequently involve using loops to iterate over large datasets. Lists are well-suited for these loop operations because of their sequential structure, which enables efficient processing of large datasets. In the code sample below, I'll show you how to create a list:

The code above shows you how to create lists containing integers, strings, and other lists. Importantly, lists do not need to contain just one data type. You can have a single list that includes integers, strings, and more. Additionally, you can add two lists together to produce a single unified list. For example, if you were to write the code new_list = list_1 + list_2, then print(new_list), the resultant output would be as follows: [1, 2, 3, 4, 5, 'A', 'T', 'G', 'C']. Next, I'll demonstrate how to add and remove elements from a list:

In the code above, I used the .append( ) function to add a new object to my list and the .remove( ) function to remove an item. To call the append function, you type your list's name followed by a period, the word append, and open and closed parentheses without spaces. You then type the object you wish to add to the list inside the parentheses. If you're adding an integer, you type the number as is, whereas if you add a string, it must be in quotation marks. The remove function works similarly, except you type the object as it appears in your list, within the parentheses, as demonstrated above. Next, i’ll show you how to call items from a list and slice lists:

In the code above, I show you how to call items from a list, which involves writing the list name followed by square brackets and the index number of the item you wish to retrieve from the list. You’ll notice that I write list[3] in my example, but the resultant output is the letter d, which is the fourth item in my list. The reason for this is that list indexing in Python follows zero-based indexing, where the first element in a list is at index 0, the second element is at index 1, and so on.
Next, I show you how to slice lists, which is the process of extracting a portion of a given data sequence using the following notation: [start: stop: step]. This notation creates a new list with elements from the index start point up to, but not including, the stop point, with an optional step specifying the increment. For example, the code list[1:4] retrieves items in positions 1-3, whereas the code list[0:4:2] retrieves times 0-3 in increments of two, which in practice means items 0 and 2. In code line 5, in the example above, you’ll see that I failed to input a stop position for my list. This retrieves all items from my specified start index onward. Alternatively, in line 6, I failed to input a start position, which means starting at index 0 and retrieving all items unto, but not including my specified stop position.
In the following block of code I’ll show you how to loop through lists:

Looping through lists enables the repeated execution of operations across numerous data points. In general, looping through a list takes the following form:
for item in list:
# code to execute for each item In essence, the code above works because the loop processes each item in the list and then performs the specified operations on that element. Later in the article, I’ll provide a more comprehensive overview of for loops and while loops under the Control Flow section. Now, we’ll wrap this sub-section on lists up with the following demonstration on list comprehensions:

List comprehensions provide a concise and readable way to create lists and perform operations on existing ones. In bioinformatics, where data manipulation and analysis are frequent tasks, list comprehensions offer a compact syntax for tasks like filtering, transforming, or generating lists of data. For example, the list comprehension above calculates the GC content in various DNA sequences stored in a list by counting the occurrences of ‘G’ and ‘C’ and dividing that sum by the sequence length.
Working With Dictionaries:
A Python dictionary is a built-in data type that stores and organizes data in key-value pairs. In other words, it's a collection of items, each identified by a unique key, and each key is associated with a corresponding value. Keys must be strings, numbers, or tuples, and values can be of any data type, including numbers, strings, lists, or even other dictionaries. In the code block below I'll show you how to create a dictionary:

As previously stated, dictionaries excel at storing data in key-value pairs. In bioinformatics, this mirrors the relationship between biological entities and their associated attributes. For example, a dictionary can contain patient data, such as in the example above. In this case, the key is the patient's ID, and the value is another dictionary containing information about the patient. In the second example above, we have a dictionary called dna_to_rna, where each key is a DNA nucleotide, and the value is the associated RNA nucleotide. After defining a dictionary, you can retrieve information based on a specific identifier (i.e., key).
The dictionary is mutable, meaning you can modify its content by adding, removing, or updating key-value pairs. In the code below, I'll demonstrate how to add and remove key-value pairs from a dictionary:

In the code above, I use the .pop( ) function to remove a key-value pair from my dictionary. To call the pop function, you type your dictionary name followed by a period, the word pop, and open and closed parentheses without spaces. You then type the key for the key-value pair you wish to remove within the parenthesis. To add a new key-value pair to your dictionary, you write the name of your dictionary followed by close brackets and an equal sign. You then type the key for your new key-value pair within the closed brackets and the new value after the equal sign, as demonstrated in the code above.
In the final demonstration in this section i’ll show you how to convert a tuple into a dictionary:

The sample code above is a simple example of converting a list of tuples into a dictionary. However, in bioinformatics, you might encounter more complex scenarios where converting tuples to a dictionary is beneficial for better data representation, organization, and analysis, as demonstrated in the code below:

In the code above we have gene annotations provided as a list of tuples, where each tuple represents information about a gene, such as gene name, start position, end position, and gene type. We then use a list comprehension to convert the list of tuples store in gene_annotation into a dictionary called gene_dictionary.
◈ ◈ ◈
🐍 Control Flow With Conditionals And Loops 💻
Control flow is crucial in bioinformatics for navigating and processing diverse biological data by controlling the order in which statements are executed in a code block. Additionally, control flow provides the flexibility to handle different conditions, iterate over data, implement algorithms, and automate tasks, contributing to the efficiency and effectiveness of bioinformatics analyses and computational biology research.
In this section, I’ll cover the two most common types of control flow you’ll encounter: conditionals and loops. Conditionals help you make decisions in your code by executing specific blocks based on whether a condition is true or false. Loops, on the other hand, enable you to repeat a set of instructions multiple times, making it easier to process data or perform repetitive tasks.
Control Flow With Conditionals:
Conditionals in Python are structures that allow you to execute certain code blocks based on whether a specified condition is true or false. In the sample code below I’ll demonstrate the simplest type of conditional, called conditional execution:

In the code above, we have a simple example of conditional execution. If the logical statement x > 10 is true, then the code in the indented statement below it is executed. If the logical statement is not true, then the indented statement is skipped, and nothing happens. Next, I’ll demonstrate another type of conditional called alternative execution:

The fundamental logic behind alternative execution is similar to conditional execution, with the slight difference that code is executed whether or not the logic statement is true. For example, we have a logical statement if >10 in the code block above. If the logical statement is true, the indented code on line 4 is executed. Alternatively, the indented code on line 6 is executed if the logic statement is false. Once you understand alternative execution, the next type of conditional, called a nested conditional, is easy to comprehend:

A nested conditional refers to a situation where one or more conditional statements are nested inside another conditional statement. In other words, there is a conditional statement within the block of code controlled by another conditional statement. This nesting can occur at any level, creating a hierarchy of conditions. The example above shows an outer if statement that checks if x and y are equal. If that statement is true, the code prints ‘x and y are equal.’ If the outer statement is false, the else block containing another if/else alternative conditional is executed.
Now, before we review loops there is one more type of conditional to review, called a chained conditional:

Whereas a nested conditional refers to one or more conditional statements nested inside another conditional statement, a chained conditional is a conditional statement that contains a series of alternative branches using if, elif, and else statements that are all indented at the same depth, as demonstrated in the code block above.
Control Flow With Loops:
In Python, loops enable you to repeat a block of code multiple times, making it easier to process data or perform repetitive tasks. There are two types of loops you should be familiar with: for loops and while loops. A for loop iterates over a sequence (such as a list, tuple, string, or range) and executes a code block for each element in that sequence. The syntax of a for loop is as follows:
for variable in sequence:
# code to execute for each element in the sequenceIn bioinformatics, a common scenario involves iterating over a collection of biological sequences, such as DNA, RNA, or protein sequences. In the example below, I’ll show you how to use a for loop for this task:

In the example above, the code for sequence in dna_sequences: initiates the for loop. For each item (sequence) in our list, the code then calculates the length and prints the length of each DNA sequence and the sequence itself.
Whereas for loops iterates over a sequence and executes a code block for each element in that sequence, while loops repeatedly execute a block of code as long as a specified condition is true. The syntax of a while loop is as follows:
while condition:
# code to execute as long as the condition is TrueAs previously mentioned, for loops are commonly used in bioinformatics for iterating over sequences or datasets. While loops, on the other hand, are useful in scenarios where the number of iterations is not known beforehand or when iterating until a specific condition is met.
In bioinformatics, a while loop might be used to simulate a scenario where a biological process continues until a certain condition is met. Let's consider a simplified example where we simulate a mutation in a DNA sequence until a specific mutation pattern is achieved:

In the code block above, the while loop is active as long as dna_seq and target_mutation are not equal. In each iteration through the while loop, a random position in dna_seq is chosen and the base at that position is mutated. The while loop tracks the number of mutations performed until dna_seq =target_mutation.
◈ ◈ ◈
🐍 Creating Modular Code With Functions 💻
A function is a reusable block of code that performs a specific set of tasks. For example, in bioinformatics, functions are often used for tasks such as sequence manipulation, statistical analysis, or data preprocessing. Functions enhance the readability of code, allowing for the reuse of code snippets and facilitating the development of robust and modular bioinformatics workflows. The code block below provides you with the basic Python function syntax:
def function_name(parameter_1, parameter_2, ...etc.):
# code to perform a task
# optionally, return a resultThere are a few key components in the code example above. First, the word def is the keyword used to define a function in Python. Next, function_name is the function's name, which you'll use to call the function later on. Ideally, the name should be descriptive. After that, we have a set of parentheses followed by a colon. You can declare one or more parameters or inputs the function accepts within the parentheses. Then, the indented code blocks contain the statements that define what the function does. Often, functions will end with a command to return a specific value using the return statement. Below is a simple example of a function that takes a dna_sequence as its single input and then returns the corresponding rna_sequence:

In the example code above, the name of my function is dna_to_rna, and the function takes a DNA sequence as input. Inside the function, my code creates a new object named rna_sequence, which is defined by the code dna_sequence.replace('T', 'U'). My function then returns the rna_sequence using the return statement.
I then call my function with the code result = dna_to_rna(dna_seq). Importantly, when you call a function, you must write the function's name, in this case, dna_to_rna( ), and then put the input parameter within the parenthesis. You'll note that the parameter I inputted when I called the function is different than when I defined the function. This is okay as long as the input parameter I choose is formatted correctly for the operations my function performs.
I provided an example of a simple function in the code above. However, in bioinformatics, functions often call other functions to create modular and reusable code. In the code below, i'll provide an example where one function calculates the GC content of a DNA sequence, and another function uses this information to determine whether the GC content is low, moderate, or high:

In the code above, the first function, find_gc_content( ), computes the GC content of a given DNA sequence, then the second function, analyze_gc_content( ), calls the first function to get the GC content of a DNA sequence and then analyzes the result.
◈ ◈ ◈
🐍 Data Manipulation With Pandas 💻
Pandas is a powerful open-source data manipulation and analysis library for Python. It provides data structures, such as series and data frames, designed to efficiently handle and analyze structured data. Pandas is widely used in various fields, including bioinformatics, for data cleaning, exploration, and analysis tasks. In the code below I’ll show you how to import the Pandas library, how to load a CSV file as a Panda’s data frame, and how to display the data frame as a table:

I import the Pandas library in the code block above by typing import pandas as pd. I then load a CSV file with information about different viruses using Panda’s pd.read_csv( ) function, which allows me to store the CSV file as a data frame in the df variable. I then use IPython’s display( ) function to display my datagrams, resulting in the above table. You can also choose to display a specified number of the first or last set of rows using [name of data frame].head( ) and [name of data frame].tail( ), respectively. For example, if you only want to view the first ten rows of data, you can type [name of data frame].head(10). Now, let’s say that when viewing your data, you notice missing values. In that case, you can use the [name of data frame].dropna( ) command, which removes all null values from your data.
After viewing your data frame, you can perform more complex exploratory data analysis. For example, in the code below, i’ll show you how to generate summary statistics for the data in your data frame using the .describe( ) command:

After using Pandas to load a data frame, you can also use additional libraries such as matplotlib.pyplot for performing exploratory data analysis with various visualization tools. For example, in the code below I'll demonstrate how to generate a scatter plot to better understand the relationship between viruses inner and outer radius:
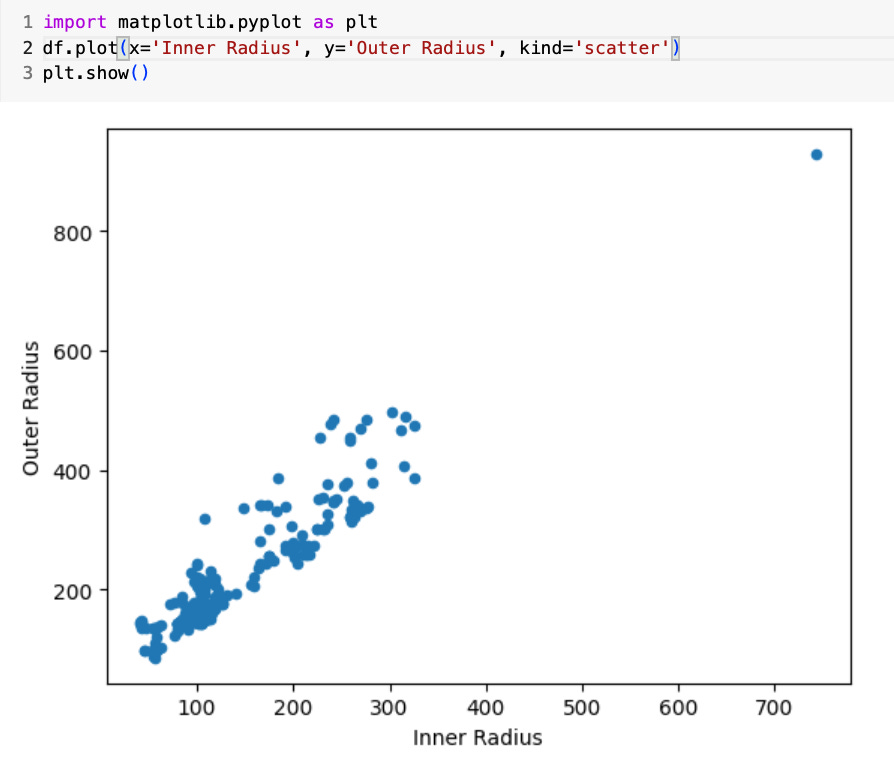
◈ ◈ ◈
Want To Learn More? Check Out The Following Related Newsletters!



Additional Resources:
DNA Confesses Data Speak is a phenomenal resource for bioinformaticians, as is the eBook From Cell Line To Command Line.
The Biostar Handbook is another great resource for all things computational biology.