



Unraveling Evolutionary Secrets through Sequence Alignment


Decoding Biology is now read in 49 US states and 91 countries. Thank you to all of my readers for your support! If you enjoy today’s newsletter, please consider tapping the ❤️ in the header above. It helps me understand which pieces you like most and supports this newsletters continued growth. Thank you!
◈ ◈ ◈
🧬 Introduction To Sequence Alignment 🧫
Sequence alignment is a fundamental technique in bioinformatics that involves arranging and comparing two or more biological sequences (DNA, RNA, or proteins) to identify similarities, differences, and patterns within the data. Sequence alignment serves several important purposes, including homology detection, which can help determine evolutionary relationships between organisms by identifying conversed DNA regions suggesting shared ancestry. Additionally, sequence alignment can be used to compare sequences from the same species to identify mutations or gene variations associated with diseases. In this newsletter, we’ll explore sequence alignment and its importance, the differences between global and local sequence alignment, commonly used sequence alignment algorithms, and ways for you to easily harness the power of programming languages to perform sequence alignment.
◈ ◈ ◈
🧬 Pairwise Sequence Alignment 🧫
Pairwise sequence alignment is a bioinformatics technique used to compare two biological sequences (usually DNA, RNA, or protein sequences) and identify regions of similarity or dissimilarity between them, often with the aim of determining evolutionary relationships between different organisms. This is possible because sequences are related through descent with modification, and related molecules have similar functions in different organisms, as demonstrated in the image below.

The primary goal of pairwise sequence alignment is to find the best-matching piecewise alignment of two query searches by inserting gaps, mismatches, and matches to maximize the similarity or identity score. Pairwise alignments can only be used between two sequences at a time, but they are efficient to calculate and are often used for methods that do not require extreme precision (such as searching a database for sequences with high similarity to a query).
Scoring alignments involves assigning scores or penalties to matches, mismatches, gaps, and other alignment features. Before we discuss specific scoring schemes it is important to understand the various (dis)alignment features and why they may occur. Let’s use the following two nucleotide sequences as an example:

The first source of dis-alignment we'll review is mutation, which is when a nucleotide in a specific location is replaced by a different nucleotide. For example, in sequence 1 (featured above), the first three nucleotides are 'ATC,' but a mutation in the third nucleotide results in 'ATG.' Additionally, a mutation in the fourth nucleotide, 'G,' results in it being replaced with an 'A'. Generally, consecutive mutations are more likely to occur in DNA sequences that are less crucial for an organism's survival (otherwise, the organism wouldn't reproduce and pass on said mutations).
The next source of dis-alignment is an insertion, which is when a nucleotide is inserted between two existing nucleotides. For example, if the starting sequence was 'AC' and guanine was inserted between 'A' and 'C,' the resulting sequence would be 'AGC'. On the converse, we can have a deletion, which is when a nucleotide is removed from a sequence. Collectively, insertions and deletions are called indels, which single mutation events, retrovirus insertions, unequal crossover during meiosis, or translocations of DNA between chromosomes can cause.
Now, as we begin discussing different ways of scoring sequence alignment, it’s important to realize this process has a bit of an arbitrariness. For example, below, you’ll find two different ways of aligning two sequences:

Suppose we use standard alignment scoring (+1 for match, -1 for mismatch, and -2 for gap). In that case, we get alignment scores of 6 and 4 for the two solutions, respectively, indicating that solution 1 is more favorable. Below you’ll find sample code automating this process where we use the pairwise2 model and align sub-module’s globalms() function to align our two sequences. The globalms() function takes our two sequences as inputs, followed by our scoring system for matches, mismatches, gaps, and gap extensions respectively.

However, what if we used a biologically inspired substitution matrix, as demonstrated below, which penalizes some mutations much more heavily than others, according to their biological effects:

Now, suppose we score the same two solutions above using the biologically inspired substitution matrix. In that case, the resulting scores are 66 and 69, indicating that solution 2 is more favorable, which demonstrates the fact that the specific scoring system you use for sequence alignment determines which of your proposed solutions are deemed optimal.
◈ ◈ ◈
🧬 Global Vs. Local Alignment 🧫
In the previous section, I demonstrated how very short and similar sequences can be aligned by hand. However, most real-world sequence alignment problems require the alignment of highly variable and lengthy sequences, which cannot feasibly be done manually. Instead, algorithms produce high-quality sequence alignments, and human ingenuity is occasionally required to make final tweaks to the aligned sequences. Computational or algorithmic approaches to sequence alignment typically fall into two categories: global and local. Global alignment is a form of 'global optimization' that forces the alignment to span the entire length of the queried sequences. In contrast, local alignment identifies regions of similarity within queried sequences. For example, in the code below I’ll demonstrate a local alignment using Biopython’s localms() function:

In global alignment, the algorithm tries to find the best alignment of the complete sequences, and it typically includes matches, mismatches, gaps, and extensions at both ends of the sequences. Global alignments are useful for identifying homologous genes or proteins, comparing closely related sequences, and aligning complete sequences for phylogenetic analysis. Additionally, in global alignments, you usually apply linear gap penalties to account for gaps in the entire sequence, which encourages longer gaps. The Needleman-Wunsch algorithm is an example of an algorithm used for global alignment.
Needleman-Wunsch is a global alignment algorithm used for aligning two sequences. It uses dynamic programming to find the optimal alignment based on the scoring scheme. It guarantees finding the best alignment but is computationally intensive for large sequences.
Local alignment, on the other hand, focuses on identifying the most similar subsequences within the two input sequences, often with the goal of identifying functional domains, motifs, or binding sites. Local alignment aims to find regions of high similarity, ignoring the regions of dissimilarity at the sequence ends. Additionally, local alignments often use affine gap penalties, where the gap opening and extension penalties differ. This encourages the alignment of shorter, high-scoring regions within sequences. The Smith-Waterman algorithm is a widely used method for local alignment.
Smith-Waterman is a local alignment algorithm based on dynamic programming. It's used to find the most significant local alignment between two sequences, allowing the identification of specific domains or regions of similarity.
The choice between global and local sequence alignments depends on the specific biological or computational question you are trying to address. Global alignment is suitable for comparing entire sequences to determine overall sequence similarity. In contrast, local alignment is better for finding specific conserved regions or similarities within sequences, which can be particularly useful in identifying functional domains or motifs in proteins and genes. Additionally, alignment tools, such as BLAST, offer efficient ways to perform alignments on vast databases.
BLAST is a popular sequence alignment algorithm used for searching large sequence databases efficiently. It combines heuristics and dynamic programming to find local alignments quickly. It's highly versatile, used for comparing sequences of various types (DNA, RNA, proteins) and searching against extensive databases.
◈ ◈ ◈
🧬 Sequence Alignment In Biopython 🧫
Evolutionary relationships among species are often inferred by comparing genetic sequences, and sequence alignment is a common method for assessing similarities and differences between homologous genes.
The leptin gene provides instructions for making the hormone leptin, which regulates food intake and energy utilization in response to the adequacy of body fat reserves. Additionally, the leptin gene is shared by humans, chimpanzees, and mice, suggesting a common ancestor for these three species. However, it shouldn’t be surprising that based on sequence alignment, the nucleotide sequences coding for the leptin gene in humans and chimpanzees are more similar than those in humans and mice, suggesting that humans and chimpanzees are more closely related.
In the sample code below, i’ll perform global sequence alignment for the first one hundred nucleotides in the human, chimpanzee, and mouse leptin gene:

As you can see, with standard alignment scoring, the match between the human and chimpanzee DNA sequences is significantly higher than the human and mouse DNA sequences or chimpanzee and mouse sequences. When aligning the nucleotide sequences for the entire leptin gene, this becomes even more exaggerated, with the alignment score for the human and chimpanzee sequences reaching ~14,500 and the human and mouse sequences reaching ~ -1,000 (the negative score is due to the high number of gap penalties). If you were to perform this same type of alignment for enough different organisms, you can begin to construct a detailed phylogenetic tree for the leptin gene, as demonstrated in the image below:
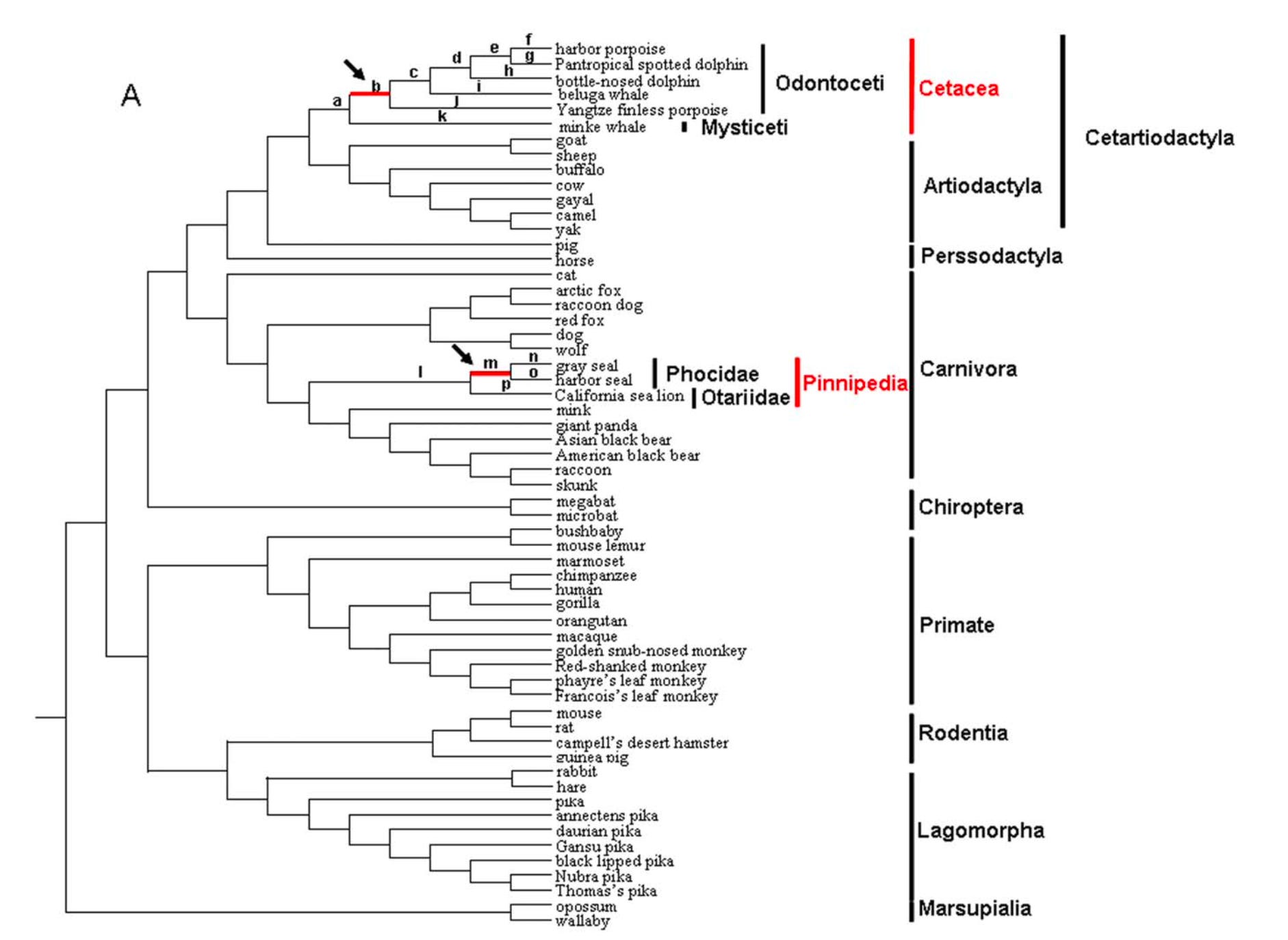
This type of analysis is possible because when the sequences of leptin genes for many organisms are compared, the degree of similarity or divergence can be determined. For example, using the code above, we can see that the human and chimpanzee leptin gene sequences are more similar to each other than either is to the mouse sequence, implying a closer evolutionary relationship between humans and chimpanzees. The reason for this is that genetic sequences accumulate mutations over time, and thus the more mismatches between two homologs, the longer the amount of time that has elapsed since they shared a common ancestor.
◈ ◈ ◈


