



An Introduction To Genome Reference Assemblies
Intro to GENCODE, genome reference assemblies, and GTF file types.


🧬 What Is GENCODE and What Are Genome Reference Assemblies?

GENCODE is a project and database focusing on the comprehensive annotation and analysis of the human and mouse genomes. The goal of the GENCODE project is to identify, classify, and understand all of the functional elements within human and mouse genomes. Additionally, GENCODE released all their annotations for human and mouse reference genomes to benefit the biomedical research community. In this article, I'll be working with the data from GRCh38, the latest human reference genome at the time of my writing. You can download GRCh38 here.
GRCh38, or Genome Reference Consortium Human Build 38, is a specific version of the human genome reference assembly. It represents the 38th major version of the reference genome for the human species. Genome assemblies are comprehensive representations of the DNA sequences and genetic content of a specific organism, and they serve as a fundamental resource for genetic and genomic research.
Genome assemblies do not typically represent one specific individual. Instead, they are typically a composite representation of a species or population. This composite representation is created by combining and averaging the genetic material from multiple individuals of the same species.
Genome assemblies like GRCh38 are used in various biological and medical research, including identifying genes, regulatory elements, and genetic variations. They are vital for understanding human genetics, studying diseases, and interpreting genomic data from various sources, including high-throughput sequencing technologies.
🧬 How Are Genome Assemblies Made?
Genome assemblies are comprehensive representations of an organism's entire DNA sequence, including its chromosomes, genes, non-coding regions, and structural elements. They serve as reference maps that provide a complete description of the genetic content of a species, which serve as valuable resources for understanding an organism's genetics, evolution, and biolog. In general, irrespective of the sequencing technology you choose, the same multi-step process is used to create a genome assembly, as summarized below:
Sequencing: The first step in genome assembly is to sequence the organism's DNA. This is typically done using high-throughput sequencing technologies, such as next-generation sequencing (NGS). The DNA is broken into small fragments, and each fragment is sequenced independently.
Preprocessing: After sequencing, the raw data generated from the sequencers undergoes preprocessing to remove low-quality reads, correct errors, and filter out artifacts. This step ensures that the data used for assembly is of high quality.
Sequence Alignment: The next step is to align the sequenced reads to a reference genome if available. This is common in resequencing projects where the aim is to compare a new individual's genome to a well-characterized reference genome. However, for de novo genome assembly (creating a genome assembly from scratch), this step is omitted.
Annotation: The final genome assembly is annotated to identify genes, non-coding elements, and regulatory regions. Annotation involves the prediction of protein-coding genes, identification of structural features, and functional annotation of genes.
Comparison and Validation: The assembled genome is often compared to other related genomes, and its accuracy is validated through experimental methods and functional analyses.
The creation of a genome assembly is a complex and iterative process, and the quality of the assembly depends on factors such as the quality of sequencing data, the complexity of the genome, and the assembly algorithms used. In general, irrespective of the sequencing technology you choose, the workflow is similar. Additionally, the aim is generally to create a genome assembly with the longest possible assembled sequences, the least fragmentation, and the smallest number of misassembles.
🧬 Working With Genome Annotation Data
To work with genome annotation data, the GTF files you download from GENCODE need to be parsed and organized into a structured format that can be used for downstream analysis and visualization. It's common to work with such annotation files in genomics to understand the functional elements and features of a genome. The sample code, demonstrated below, efficiently extracts and organizes this information for further investigation:
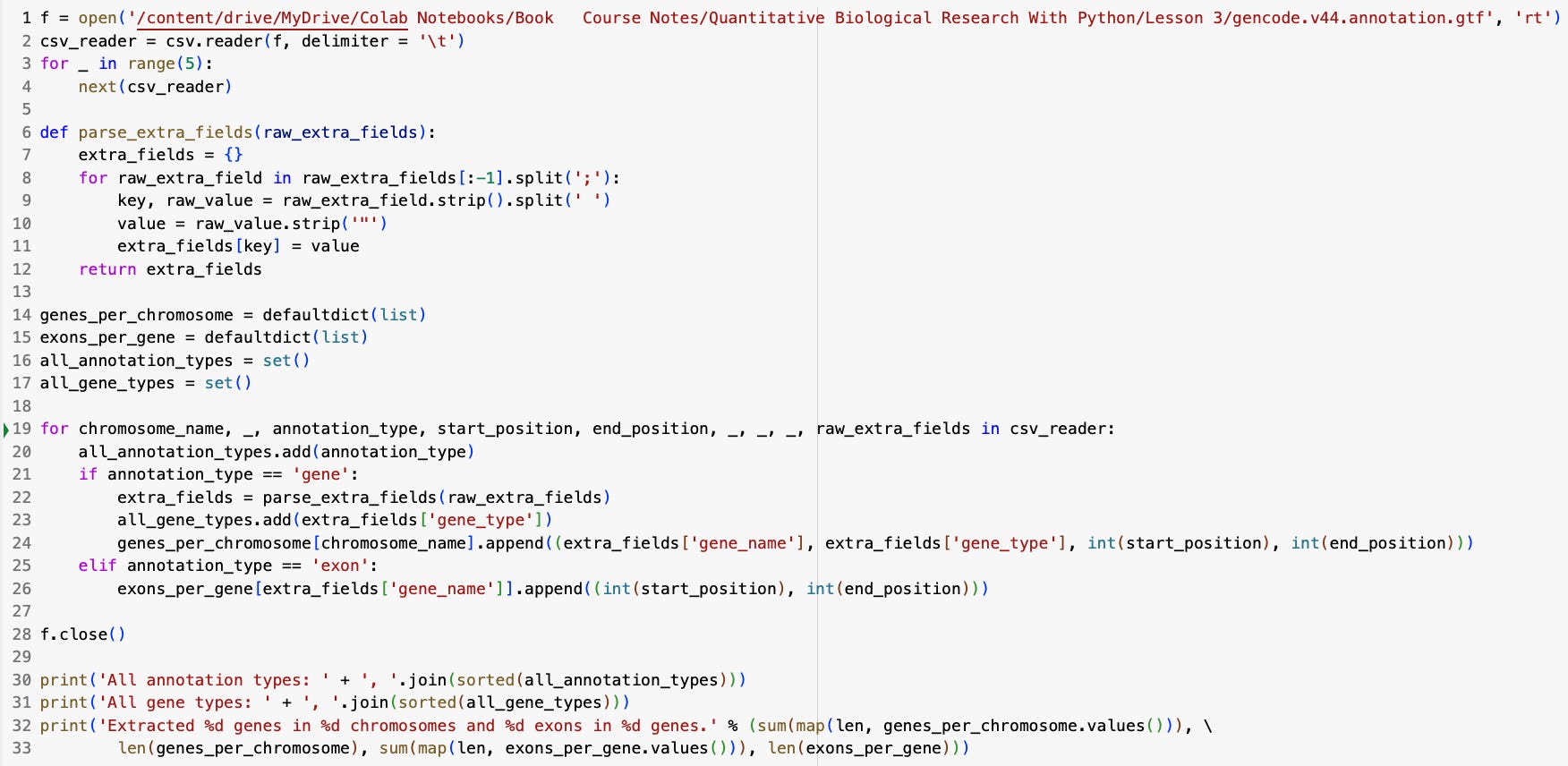
Next we’ll break this code down section by section to explain how it works:
f = open(....etc.)
csv_reader = csv.reader(f, delimiter = '\t')
for _ in range(5):
next(csv_reader)Collectively, this first code block parses our annotation file in GTF format from the GENCODE database.
The code f=open(…etc.) opens a file named gencode.v44.annotation.gtf located at the specified path in ‘rt’ (read text) mode.
csv_reader=csv.reader(f, delimiter=’\t’) creates a CSV reader object named csv_reader, using a tab (‘\t’) as the delimiter. Since GTF files are tab-delimited, the previous code helps parse the file correctly.
Then, a for loop is used to skip the first four (range(5)) lines of the GTF file, which contains header/metadata versus actual annotation data.
def parse_extra_fields(raw_extra_fields):
extra_fields = {}
for raw_extra_field in raw_extra_fields[:-1].split(';'):
key, raw_value = raw_extra_field.strip().split(' ')
value = raw_value.strip('"')
extra_fields[key] = value
return extra_fieldsThe code above defines a function called parse_extra_fields, which extracts key-value pairs from a string of raw extra fields in our GTF file.
First, we define our function with the following code, def parse_extra_fields(etc.), which takes a single argument, raw_extra_fields. Inside the function, an empty dictionary named extra_fields is initialized, which will later store the extracted key-value pairs from the raw_extra_fields string.
Then, a for loop iterates through the semicolon-separated parts of the raw_extra_fields string by splitting the string into parts using split(';'). This creates a list of substrings, each representing a single field in the extra fields section. The field is assumed to be in the format of a key-value, where the key is the string and the value is a string enclosed in double quotes, with a space between the two parts.
The code key, raw_value = raw_extra_field.strip().split('') uses the strip() method to remove whitespace from the field and the split('') method to split the field into two parts (the key and raw value). This assumes a space character between the key and raw value.
Next, the code value = raw_value.strip("") extracts the raw values and removes the double quotes surrounding them (values in GTF files are often enclosed in double quotes). The key-value pair is then added to the extra_fields dictionary, where the key is associated with the extracted value.
Finally, after processing all the fields in raw_extra_fields, the extra_fields dictionary contains all key-value pairs extracted from the input string. Then, the extra_fields dictionary is returned from the function, allowing you to access specific values by their keys.
genes_per_chromosome = defaultdict(list)
exons_per_gene = defaultdict(list)
all_annotation_types = set()
all_gene_types = set()First, this code creates a default dictionary called genes_per_chromosome that stores gene information using chromosome names as keys and genes as values. Importantly, each stored gene is a tuple with four elements: gene_name (str), gene_type (str), start_position (int), and end_position (int). Next, a second dictionary called exon_per_gene is created, which stores exon information using chromosome names as keys and exons as values as tuples containing start and end positions (start_position and end_position) for each chromosome. Finally, two sets are created. First, all_annotation_types is an empty set that will later contain all of the annotation types in the GTF file, and second all_gene_types is an empty set that will later contain all of the gene types in the GTF file.
for chromosome_name, _, annotation_type, start_position, end_position, _, _, _, raw_extra_fields in csv_reader:
all_annotation_types.add(annotation_type)
if annotation_type == 'gene':
extra_fields = parse_extra_fields(raw_extra_fields)
all_gene_types.add(extra_fields['gene_type']) genes_per_chromosome[chromosome_name].append((extra_fields['gene_name'],extra_fields['gene_type'], int(start_position), int(end_position)))
elif annotation_type == 'exon':
exons_per_gene[extra_fields['gene_name']].append((int(start_position), int(end_position)))
f.close()First, the code block above starts with a for loop, which reads and processes the main annotation data in the GTF File by iterating over each line of the file and extracting information such as the chromosome_name, annotation_type, start_position, end_position, and raw_extra_fields.
Next, the code all_annotation_types.add(annotation_type) collects unique annotation types in the GTF file and adds them to the all_annotation_types set. After processing all of the annotation types the all_annotation_types set will contain the following elements: DS, Selenocysteine, UTR, exon, gene, start_codon, stop_codon, transcript
if annotation_type = ‘gene’: looks at the current annotation type and if it is labeled ‘gene’ then the code uses the parse_extra_fields function to extract additional information from the extra fields. The code then adds the gene type to the all_gene_types set. Following that, an entry to the genes_per_chromosome dictionary using chromosome_name as the key and appending the following as the values: extra_fields[‘gene_name’], extra_fields[‘gene_type’], int(start_position), int(end_position).
Alternatively, if annotation_type=’exon’, the code adds the exon’s start and end positions to the appropriate gene’s exon list using the exons_per_gene dictionary. Finally, after processing all of the annotation lines, the file is closed using the f.close() statement.
print('All annotation types: ' + ', '.join(sorted(all_annotation_types)))
print('*' * 50)
print('All gene types: ' + ', '.join(sorted(all_gene_types)))
print('*' * 50)
print('Extracted %d genes in %d chromosomes and %d exons in %d genes.' % (sum(map(len, genes_per_chromosome.values())), \
len(genes_per_chromosome), sum(map(len, exons_per_gene.values())), len(exons_per_gene)))Finally, the code above prints summary information including all unique annotation types, all unique gene types, and the total number of genes and exons extracted from the file, resulting in the following output:

The code output above provides an overview of the different annotation and gene types present in the annotation file, as well as statistics on the number of genes, chromosomes, and exons extracted from the file. Now, let’s analyze each of these three sections:
All annotation types list the unique annotation types found in the annotation file, which represent the different features in the genome. For example, “CSD” refers to coding sequences, and each subsequent comma-separated item is a different annotation type.
All gene types list the unique gene types in the annotation gene, where the listed types refer to different functional or structural categories of genes. For example, “lncRNA” represents long non-coding RNAs, “miRNA” stands for microRNAs, and so forth.
Extracted 62700 genes in 25 chromosomes and 169476 exons in 61228 genes summarize the data extracted from the annotation file. The number of chromosomes includes the 22 autosomes, the X and Y chromosomes, and mitochondrial DNA (listed as M chromosome in the GTF file), which is why the count is greater than 23. Additionally, the number of unique genes seems extraordinarily high, which is explained by the fact that some genes have multiple isoforms or annotations in the GTF file.
🧬 Quantifying Protein Coding and miRNA Genes
As previously demonstrated, there are 61,228 genes identified in human genome assembly GRCh38. However, the distribution of these genes is not uniform across chromosomes. By quantifying the number of protein-coding genes on each chromosome, we can pinpoint candidate genes responsible for specific conditions. Additionally, since micro RNAs play a crucial role in regulating coding genes by interacting with coding genes and binding their mRNAs to influence translation or stability, understanding the distribution of miRNA across chromosomes can help us better understand their influence on the expression of coding genes.
An analysis of the human genome assembly shows there are 20,336 protein-coding genes and 3030 miRNA genes. Because we’ve already 169476 exons, this means that every protein-coding gene in the genome is composed of one or more exons (non-coding micro RNA also have exons but do not code for proteins). In the code sample below, I’ll show you how to determine the total number of genes, protein-coding genes, and miRNA genes on each chromosome:

Now, let’s break the code down one step at a time….
def filter_genes_of_type(genes, gene_type):
return [gene for gene in genes if gene[1] == gene_type]The code above creates a function called filter_genes_of_type, which takes a list of genes and a specific type of gene (gene_type) as the inputs. The function then returns a list containing only the genes with the specified gene type.
protein_coding_genes_per_chromosome = {}
for chromosome, genes in sorted(genes_per_chromosome.items()):
protein_coding_genes = filter_genes_of_type(genes, 'protein_coding')
miRNA_genes = filter_genes_of_type(genes, 'miRNA')
protein_coding_genes_per_chromosome[chromosome]=protein_coding_genes
print('%s: %d total genes, of which %d are protein coding and %d are miRNA genes' %(chromosome, len(genes),len(protein_coding_genes), len(miRNA_genes)))First, protein_coding_genes_per_chromosome={} initializes a dictionary that will store information about protein coding genes for each chromosome. Next, a for loop is created, which iterates over the chromosomes (keys) and their associated genes (values) stores in the genes_per_chromosome dictionary. Then, within the for loop, the code protein_coding_genes = filter_genes_of_type(genes, ‘protein_coding’) filters the genes for the current chromosome using the previously defined filter_genes_of_type function. Importantly, protein_coding_genes will only contains genes with the type ‘protein_coding’. After that, miRNA_genes = filter_genes_of_type(genes, ‘miRNA’) performs the same operation, but for miRNA genes instead of protein coding genes. Then, the code protein_coding_genes_per_chromosome[chromosome]-protein_coding_genes stores the filtered protein coding genes for the current chromosome in the protein_coding_genes_per_chromosome dictionary, and finally a print statement is used to print the output of the code, which includes the total number of genes on each chromosome, the number or protein coding genes, and the number of miRNA genes.
I’ve summarized the output of the code above in the chart below:

In the image above we can see the total number of genes, proton-coding genes, and miRNA genes on each chromosome, which conforms with the data presented in the National Center for Biotechnology Information’s chromosome map.


