



The Wide World Of Biological Data Formats
Navigating common bioinformatics data formats, naming schemes, and databases


🔬The Wide World Of Biological Data Formats
One of my biggest challenges getting into bioinformatics was navigating the diverse world of data formats and file types, seemingly unorganized naming conventions, and the idiosyncrasies of specific databases and data sources. The countless hours I spent scouring the web for information led me to a realization: even seasoned scientists in the field grapple with these data-related challenges1. As a result, I wanted made this guide to teach you about the different types of data formats you’ll encounter in bioinformatics and what their similarities and differences are.
🔬What Is Data and What Are Data Formats?
There are so many data formats and file types used in bioinformatics. Superficially, it would seem that having a single standardized data format for bioinformatics analyses would make everyone's lives easier. However, this bandaid solution neglects the fundamental reason why we have different data types and formats in the first place.
Data is a symbolic representation of information, but its interpretation hinges on the chosen format. Data formats are systematic designs aimed at enhancing the comprehensibility of information. Unlike a one-size-fits-all approach, there isn't a singular data format that perfectly captures a set of information. This isn't dissimilar to what we see in the image below, where a flashlight illuminates the same cube from three angles, producing three very different shadows. In the same way, three different data formats can be applied to the same information, resulting in three different ways of viewing the data and three different interpretations.

The same information can be articulated through various data formats, each excelling in distinct aspects of conveying the information. At times, certain information within data remains elusive or challenging to grasp due to the format employed. In these cases, the data format might not be optimized for a specific analytical task. Predicting the novel insights achievable from a dataset can be tricky, prompting a continuous quest to explore innovative approaches that stretch the boundaries of information extraction. Therefore, mastering the understanding of data formats, discerning the information embedded in each, and determining the apt format for a given situation is a fundamental skill for a bioinformatician.
🔬 Common Bioinformatics Data Formats
Data formats are ways of structuring data to enhance the comprehensibility of information. Different data formats may contain the same raw information but optimize different aspects, making one format better than another for certain goals or analyses. Data optimization is a somewhat nebulous and abstract topic without specific examples, so rather than spending additional time on the relative utility of different data formats, I’d like to dive into examples of common data formats you’ll encounter. In the subsections below, I’ll explain the three most common categories of data formats, including reference, sequence, and results formats, and the various specific data formats that fit into each of these categories.
Reference Data Formats
One of the most common types of data you’ll encounter in bioinformatics is reference data, which serves as a repositories for existing knowledge. Generally, you’ll access reference data files from databases including GenBank, the sequence read archive (SRA), or any number of project specific databases. Every year the Journal of Nucleic Acids Research publishes a database issue, which provides up to date information on the major databases used by researchers in the field. You can find the most recent issue here. The most common reference data formats include GenBank, FASTA, GFF, GTF, and BED files formats, each tailored with specific optimizations.
GenBank Files:
The GenBank file format is one of the oldest bioinformatics data formats, and it is optimized for readability by humans. However, for this same reason, GenBank files are not optimized for any particular data analysis task.
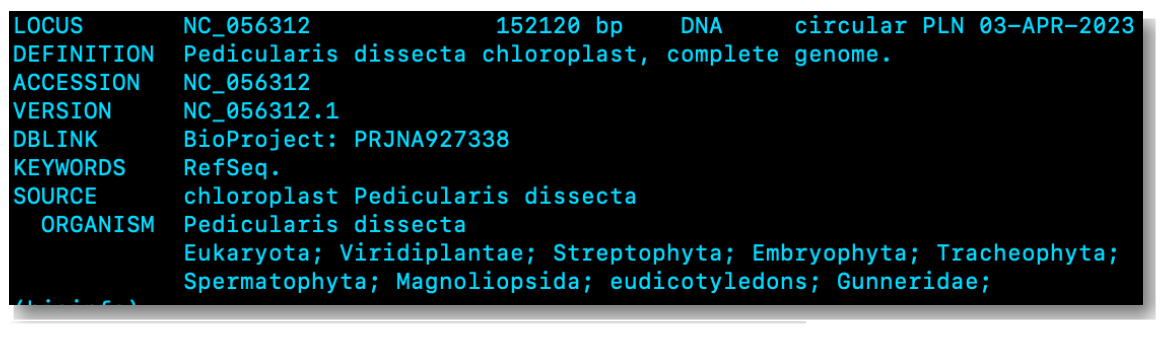
GenBank files exhibit a complex structure, making them less commonly utilized in bioinformatics analyses due to limited compatibility with bioinformatics tools. As a result, we typically convert GenBank files to another, simpler format, such as FASTA or GFF. This is only possible because GenBank files are more data-rich than FASTA or GFF files. If you were to convert a FASTA or GFF file to a GenBank file you’d more than likely end up with an incomplete file. In the code block below, i’ll show you how to obtain a GenBank file in your terminal:
bio fetch NC_056312 > genome.gb
cat genome.gb | headThe code above uses the bio fetch command to obtain data from GenBank for the specified accession number (NC_056312) and then redirects the output to a new GenBank file named genome.gb. The code then uses Unix’s cat command to view the contents of the genome.gb file and the | head portion of the code is a command-line feature that limits the output to the first few lines of data in the file. The resulting output is as follows:
FASTA Files:
FASTA files are one of the most prevalent reference data formats used in bioinformatics due to their simplicity and readability. FASTA files use a text-based format to represent DNA, RNA, and protein sequences and can even be written manually. Each entry within a FASTA file includes a header with a sequence ID and description followed by a sequence. Additionally, FASTA files are denoted by the following extensions: .fa, .fasta, .fna, and .seq. In the code block below, I’ll show you how to obtain a FASTA file from GenBank in your terminal:
bio fetch NC_056312 --format fasta > genome.fasta
cat genome.fasta | head -3The code above uses the bio fetch command to obtain data from GenBank for the specified accession number (NC_056312). However, instead of obtaining a GenBank file, the snippet of code --format fasta specifies the file format as a FASTA before redirecting the output to a new file named genome.fasta. We can then view the first few lines of this new file with the command cat genome.fasta | head -3, resulting in the following output:

In the example above, I obtained a file from GenBank in a FASTA format. However, suppose you know the accession number of a sequence deposited in NCBI. In that case, you can obtain it with the following command-line code (you should input your actual accession number, for example, NM_12000, providing you with sequence information on the Arabidopsis thaliana MATE efflux family protein):
efetch -db nuccore -id NM_000000 -format fasta | headIf you’re interested in learning basic Unix skills, you can check out the following article titled, Unix Fundamentals For Bioinformatics.
GFF and GTF Files:
In addition to GenBank and FASTA, other common reference data formats include GFF and GTF files, which encapsulate information such as the reference ID for an annotated DNA, RNA, or protein sequence, feature types, genomic coordinates (start/end positions), confidence scores, strand orientations (+/-), and data sources. Because GFF and GTF files contain information about the position of sequences, they are commonly referred to as interval-format files2. In the code block below, I’ll show you how to obtain a GFF file from GenBank in your terminal (the same concept can be applied to obtaining GTF files):
bio fetch NC_056312 --format gff > genome.gff
cat genome.gff | head -6The same basic commands used to obtain a FASTA file from GenBank can be repurposed to retrieve a GFF or GTF file. As you can see below, the same information can be conveyred in different ways based on the type of file format used, which recapitulates the point made earlier in this article.
Sequencing Reads Formats
Sequencing reads file formats are standardized ways of representing the output data generated by high-throughput DNA sequencing technologies. These files contain information about the sequences of nucleotides obtained during the sequencing process and quality scores associated with each base call. The sequencing reads file format you’ll most likely encounter is FASTQ.
FASTQ Files:
In next-generation sequencing, DNA or RNA samples undergo digitization, progressing through various steps to produce FASTQ files, as described at length in a previous article titled From Biomolecules to Bytes. FASTQ files can be thought of as an extension of the FASTA format where each sequence base is associated with a quality score (i.e., the probability that we are wrong), offering a comprehensive format for storing experimental results generated by next-generation sequencing instruments such as Illumina sequencing systems. Additionally, FASTQ files can be used as input for many different secondary data analyses. Now, in the code below, I’ll show you how to download single-cell RNA sequencing data for a bumble bee (Bombus terricola), which can be used for transcriptomics studies.
# Unix/Bash
conda install -c bioconda sra-tools
fastq-dump SRR14567205The code above first uses the conda package manager to install the SRA toolkit (sra-tools) from the Bioconda channel. Then, the fastq-dump command from the SRA toolkit is used to download the FASTQ file associated with an accession number (SRR14567205)from NCBI’s sequence read archive (SRA). The fastq-dump tool specifically converts SRA data into the FASTQ format. After using fastq-dump to download your data, you can read and analyze the resulting FASTQ file using various bioinformatics tools or programming languages. In the next code block below, I’ll show you how to use Python to read and print the sequences in our FASTQ file.
# Python
pip install biopython
from Bio import SeqIO
fastq_file = 'SRR14567205.fastq'
for record in SeqIO.parse(fastq_file, 'fastq'):
print(f"ID: {record.id}")
print(f"Sequence: {record.seq}")
print(f"Quality: {record.letter_annotations['phred_quality']}")
print("\n---\n")
python read_fastq.pyIn the code above, I first use pip to install the biopython library before importing the SeqIO module from biopython, which handles sequence files.
After installing the requisite libraries, the code code fastq_file = 'SRR14567205.fastq' specifies the path to our FASTQ file containing single-cell RNA sequencing data. Then, a for loop is used to iterate over each record (i.e., sequence entry) in the FASTQ file using SeqIO.parse(), which is a function that reads sequence data as SeqRecord objects. SeqIO.parse() expects two arguments: the first argument is a handle to read the data from (i.e., fastq_file), and the second is a lowercase string specifying the file type (i.e., fastq). For each record in our FASTQ file, the for loop prints the record ID, sequence, and quality information.
Finally, the command read_fastq.py is used to run the above Python script in the Bash shell, assuming that the script is saved in the same directory where it is executed. The image below depicts the first record printed after executing the script.

As you can see, the record in the image above contains a sequence ID, a DNA sequence, and a corresponding quality score. The quality scores above are logarithmically linked to error probabilities. For example, a score of 10 gives a 1/10 chance of an incorrect base call, 20 gives a 1/100 chance, 30 gives a 1/1,000 chance, 40 is a 1/10,000 chance, and so forth. However, it’s more common that records in FASTQ files take the following format, demonstrated below.
@SRR00000001 # Sequence ID
CAGTATCGATCAAATAGGCATTGCA # Nucleotide sequence
+
!+'CCF>ABA***<=<IHDFA##(HH # Quality scoresThe first line above, starting with the @ symbol, contains a sequence ID. Then, the second line includes the measured sequence. Next, the third line contains a + symbol, and following that, we have a quality score that is encoded in a series of symbols that appear to be written in the Zapf Dingbat typeface. These encoded numerical values each represent a Phred score, which measures the quality of identifying a given nucleotide generated from DNA sequencing. Thankfully, there are lookup tables, like the one below, for decoding numerical quality scores from the symbols used.

Results Formats
Results file formats in bioinformatics are standardized formats that store and represent the outcomes of various analyses and experiments. These formats capture the results of computational analyses, experimental measurements, and other bioinformatics processes. Some common results file formats include SAM/BAM, VCF, and CSV.
SAM/BAM Files:
SAM (sequence alignment map) and BAM (binary alignment map) are two common results format files used in bioinformatics. Both file types are typically generated by aligning next-generation sequencing data, stored in a FASTQ format, to a reference genome in a FASTA format, often using alignment tools such as BWA or Bowtie2. The resulting SAM/BAM files from this process detail all the individual pairwise alignments uncovered. The code block below shows you how to generate BAM files from an input FASTA and FASTQ file:
bwa mem reference_genome.fasta NGS_output.fastq > output.bamThe Unix code above demonstrated how to use the Burrows-Wheeler Aligner (BWA) tool for aligning short DNA sequences to a reference genome. First, the bwa mem command is used to invoke the BWA tool used for high-throughput short-read alignment. Then, the names of the input files used by the BWA tool are written. The first input file is a FASTA file (reference_genome.fasta) containing a reference genome, and the second input file is a FASTQ file (NGS_output.fastq)containing sequencing data with corresponding quality scores. The bwa mem command takes these two files as inputs; then the > operator redirects the output from this code to a BAM file named output.bam, a standard format for storing read alignments against a reference genome. The resulting output file can then be further processed or analyzed using other bioinformatics tools.
VCF Files:
VCF (Variant Call Format) files are a standard file format used in bioinformatics to represent genomic variations, such as single nucleotide polymorphisms (SNPs) and insertions/deletions (indels). These files capture information about variations observed in individual genomes when compared to a reference genome, as demonstrated in the image below.

VCF files are commonly used for storing and sharing genetic variant data and are often created from SAM/BAM files described above. To learn more about the process of variant calling to identify genomic variations from the aligned reads in a SAM/BAM file, you can check out my previous article, A DIY Guide To Genomic Variant Analysis.
🔬 Want To Learn More? Check Out The Following Related Newsletters!



The origins of these challenges, it seems, can be chalked up to a combination of local optimizations, ad-hoc naming conventions, and path dependence. It's crucial to note that this isn't a criticism, as hindsight often provides clarity. The reality is that standardizing data formats, file types, naming conventions, and database organization is a heroic task unlikely to be undertaken anytime soon. Why? Standardization requires a massive coordinated effort by many people, and there is little to no professional or financial incentive for anyone to participate in this undertaking.
BED files adhere to a similar formatting convention as GTF and GFF files, but differ slightly in that they are optimized for seamless integration with genome browsers, facilitating the visualization of genomic data
Thank you for this