



From Protein Folds to Protein Folding
How reductionism, prediction, and visualization have shaped our understanding of protein's three-dimensional structures and how they arrive in those configurations.


Liked this piece? Show your support by tapping the “heart” ❤️ in the header above. It’s a small gesture that goes a long way in helping me understand what you value and in growing this newsletter. Thanks so much!
When AlphaFold2 was first released I, like many you, was blown away. But, after the initial hype cycle died down I found myself more and more underwhelmed. This feeling of underwhelm had less to do with DeepMind’s achievement in predicting protein folds (ie, a proteins final 3D structure), and more to do with the way it was discussed in science media. While writer’s whose work I deeply admire celebrated AlphaFold2— elevating it to a paradigm-shifting technology— I found myself questioning its real-world impact.
Like many other protein structure prediction tools, AlphaFold2, and its successor, could tell us what a protein looks like in three-dimensional space. But— crucially— it can’t tell us how it arrived at that configuration. What AlphaFold2 accomplished is, of course, a major scientific milestone, but it also misses the point to some degree.
With all of the hype surrounding AlphaFold and other functionally-related tools that predict proteins folds it’s easy to forget that what we really care about is protein folding — the dynamic process where a linear of amino acids transforms into a functional 3D protein structure. This distinction between folds (the end result) and folding (the process) isn’t just semantic; it reflects a fundamental difference in how we tackle one of biology’s most complex puzzles. That is, how does a sequence of amino acids reliably fold into a specific, and precise, three-dimensional structure?
This question has captivated scientists for decades not only for its intellectual challenge, but also its practical applications in drug development, protein engineering, and understanding the evolution of complex life itself. Notably, while this problem has been worked on for decades, advancements in this field haven’t followed a straight path from A to B. The approaches used to solve the aptly named "protein folding problem" have changed dramatically over time, reflecting both conceptual shifts in how we think about the problem as well as technological advances.
As time has passed, simple theoretical models have given way to predictive algorithms which are themselves poised for disruption by advances in super-resolution microscopy, allowing for the direct visualization of real-time protein dynamics. This progression from reductionism to prediction to direct observation parallels the broader history of the biological sciences themselves — a central theme of this piece. Let’s jump in!

Understanding Through Simplification
The Lattice Model — A First Step To Understanding Protein Folds
Some of the earliest attempts to understand protein folding relied of simplification. For example, in the early 1960’s and 70’s, scientists developed models that stripped proteins down to their most basic and essential features, allowing them to create abstractions that could be analyzed mathematically. One of the most influential of these approaches was the lattice model, which aimed to elucidate the fundamental principles governing protein structure, even at the cost of molecular detail.
In the lattice model, proteins are represented as beads on a grid, where each bead represents a single amino acid and neighboring amino acids are connected in sequence— amino acid 1 connects to amino acid 2, which is connected to amino acid 3, and so on. Additionally, amino acid chains can only make 90-degree turns and beads must sit at defined points on the grid (like in the game Go), creating highly constrained representations of a protein’s structure. Below you’ll find an example lattice model for a 5-mer protein1.

Despite the lattice model’s simplicity, it has proven surprisingly useful for revealing important insights about the differences in how homopolymers and heteropolymers fold2. For example, when we examine how a 6-mer homopolymer folds using a lattice model we find multiple configurations with identical energy states, as demonstrated below. This means that no single configuration is energetically favored over the other, and that no "native structure" exists.

To understand why, we need to appreciate that all amino acid-amino acid contacts in a homopolymer have the same energy. Using the example above, let’s say the interaction εₕₕ has an energy of −1. On the left most model, there is a contact between bead 1 &6 and 2 &5, and thus the total energy is -2. Similarly, for the second from the left there is a contact between 1&4 and 3&6, and thus the total energy is also -2. The same applies for the remaining structures and as a result the homopolymer can adopt any of these configurations because none is more energetically favorable (ie, lower energy) than the others.
In contrast, heteropolymers with different types of monomers create an energy landscape where one specific configuration—the native structure—has the lowest energy and is therefore preferred. For example, consider a 6-mer polymer with the sequence HHPPHH, where H units (blue) and P units (green) have different interaction energies (εₕₕ=-1 and εₕₚ=εₚₕ=0).
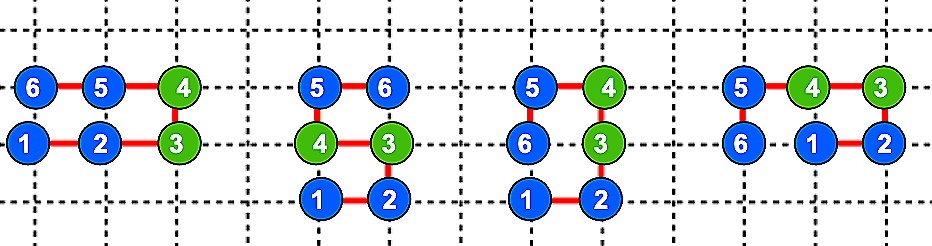
Because of the different interaction energies between the two types of monomers, the total energies vary across the above structures. For example, going from left to right the structures have total energies of -2, 0, -1, and -1. As a result, the heteropolymer selects the first structure because it has the lowest energy, representing the native state.
While these lattice models provide an overly simplified example of how proteins fold, they shine light on an important principle, which is that the diversity of amino acids in proteins create the requisite energy landscape that guides protein folding towards a defined, and specific, three dimensional structure. This concept, which came to be known as an energy funnel, is central to our understanding of how proteins find their native structures despite the nearly infinite number of possible conformations that a chain of amino acids can take on.
Beyond Lattice Models — The Molecular Mechanics Approach
While lattice models provided conceptual insights about the underlying principles governing protein structure, they lacked the detailed needed to resolve the structure of specific proteins, limiting their utility. As such, researchers quickly adopted other analytical techniques like force field models, which represent proteins as collections of atoms connected by spring with multiple energy terms representing bonds, angles. electrostatic interactions, and Van der Waals forces.

{kind=link}
By minimizing a protein’s total energy in a force field model, it became possible to predict the most stable protein configuration, which, in theory, is the native structure. However, this analytical approach faced a major limitation in that simplified force field models failed to recapitulate the complexity of the energy landscape. After all, real proteins have thousands of atoms, leading to energy landscapes with nearly limitless local minima, as depicted below.

For this reason, finding the global minimum, and by virtue of that a protein’s native structure, by energy calculation alone has proven to be too computationally expensive for all but the smallest proteins. So, again we arrive at a place where new experimental and analytical techniques yield useful insights, yet fall short of the ultimate goals of identifying a protein’s native state and how it got there — the fold and folding problems respectively.
From Principles to Structures
Knowledge-Based Predictions— Learning from Nature
Rather than building off of lattice an force field models, the next wave of experimental techniques took a conceptual leap forward— rather than attempting to derive protein structures from physical principles alone, researchers began leveraging the growing database of experimentally determined protein structures to make predictions3.
By the early 2000’s, the Protein Data Bank contained over twelve-thousand experimentally determined protein structures, providing a vast database containing information as to how amino acid sequences map to three-dimensional protein configurations, in turn enabling knowledge-based approaches to protein structure prediction like homology modeling, which identifies potential structures from a sequence of amino acids based on similarities to known proteins.
In essence, homology modeling exploits the fact that proteins with similar amino acid sequences often adopt similar three-dimensional shapes. So, if a protein with a known amino acid sequence, but unknown structure, can be aligned with a second protein where both the sequence and structure are known, then it’s possible to build a model of the unknown protein based on the known protein’s structural template. Unsurprisingly, this approach worked remarkably well for proteins with close relatives whose structures were already resolved. However, for proteins without close structural relatives it fell short, opening the door for fragment-based methods to get a foothold.
Fragment-based methods break protein sequences into short fragments, then find likely structures for each individual fragment based on similar fragments in already resolved proteins. Following that, the fragments are assembled into a complete model. The most notable example of this method is Rosetta, which is a software developed by David Baker’s lab that was used to predict the structures of multiple proteins that lacked obvious structural relatives.
Collectively, these knowledge-based methods demonstrate the power of learning from nature’s solutions to the protein fold problem rather than deriving them from scratch. However, they remained limited in their applicability because of the lack of available structural data and as a result they struggled to correctly predict the shapes of proteins with novel folds.
The Deep Learning Revolution — AlphaFold and Beyond
One of the biggest shifts in the field of protein structure prediction was the application of deep learning. Rather than relying on explicit rules or templates, deep learning algorithms identify patterns in data that might be too subtle, complex, or abstract for human recognition.

When applied to protein structure prediction, deep learning approaches achieved unprecedented accuracy with DeepMind’s AlphaFold2— which achieved near-experimental accuracy in the 2020 Critical Assessment of protein Structure Prediction competition— being a notable example. By integrating multiple sources of information including sequence conservation patterns, predicted distances between amino acids, and physical constraints, AlphaFold2 created remarkably accurate models of protein structures, even for proteins without close structural relatives.
This breakthrough transformed protein structure (ie, fold) prediction from a challenging research problem to a routine tool. The AlphaFold Protein Structure Database now contains predicted structures for over 200 million proteins, covering most known protein sequences. What once required years of experimental work can now be accomplished in minutes with computational tools — yet, we still haven’t reached the end goal.
The Critical Distinction— Folds vs. Folding
The success of predictive models like AlphaFold represents a massive achievement in determining protein folds—the final three-dimensional structures of proteins. However, it's crucial to recognize that this is not the same as understanding protein folding—the dynamic process by which proteins reach these structures.
This distinction is more than semantic. Protein fold prediction tells us what the final structure looks like, while protein folding studies reveal how that structure forms. This distinction matters for several reasons. For example, many diseases involve protein misfolding rather than misfolded proteins. The process of folding, not just the final structure, can go awry, leading to protein aggregation and cellular dysfunction. Understanding these processes is essential for developing treatments for conditions like Alzheimer's and Parkinson's disease, and prion diseases like Creutzfeldt-Jakob disease.
Additionally, creating new proteins with novel functions requires not just structures that look correct but proteins that fold efficiently and reliably. Many computationally designed proteins with promising predicted structures fail because they can’t fold properly in vivo. Finally, real-life proteins, in living cells, fold in complex environments, often with the assistance of chaperone proteins and while they're still being synthesized. These contextual factors can significantly influence the folding process and the resulting structures.
AlphaFold, and other protein structure prediction tools, effectively circumvent the folding process entirely by extracting patterns from known structures, creating a structural "parts list" that bypasses the actual process of folding. While this approach is remarkably effective for predicting folds, it tells us little about the folding process itself, which has become increasingly apparent as the field of structural biology has matures.
Collectively, scientists gained the ability to accurately predict protein structures, yet we still struggle to understand how the proteins ended up in these configurations or how changing the amino acid sequence or proteins environments impact the folding process. This gap between static structure and dynamic process points to the need for a new approach—one that could capture proteins in action rather than just their final states, which again requires a paradigm shift in how we think about the problem.
Seeing Proteins in Action
Traditional Approaches to Understanding Protein Folds and Folding
Reductionism and prediction are opposing approaches to understanding protein structure, yet both have racked up notable wins. In order to take the next leap in understanding protein folding, however, new experimental and analytical techniques are needed— is where direct visualization of protein dynamics comes in.
Rather than inferring folding pathways from computational models based on protein’s final structures, researchers are now aiming to observe the folding protects directly, capturing the movements and interactions that transform linear chains of amino acids into functional 3D protein structures.
Traditionally protein dynamics were studied using techniques like stopped-flow experiments, hydrogen-deuterium exchange, and single-molecule fluorescence, which provides valuable insights while also suffering from significant limitations. Stopped-flow experiments could only capture events on the millisecond timescale, missing faster folding events, hydrogen-deuterium exchange provided structural information wit limited temporal resolution, and single-molecule fluorescence required modification of the protein with fluorescent labels, potentially altering the very dynamics being studied.
More recent advances in nuclear magnetic resonance spectroscopy and cryo-electron microscopy have improved our ability to capture protein structures in different states but still provided snapshots rather than continuous views of protein dynamics, leaving the question — how do we observe protein folding as it happens, in real time, without disrupting the process being studied?
Tracking Single Molecules in Living Cells
One breakthrough approach to the challenge of observing protein folding in real-time has emerged from the company Eikon Therapeutics4, which has developing technologies to track individual protein molecules in living cells.
Eikon’s core technology builds on advances in single-molecule tracking microscopy, allowing researchers to follow the movements of individual protein molecules with unprecedented precision. By labeling proteins with bright stable fluorophores and using sophisticated optical systems, Eikon can track proteins in real time as they move, interact, and change conformation—including folding and unfolding events.
What makes this approach particularly powerful is its ability to operate in the natural cellular environment. Rather than studying proteins in isolation or in artificial systems, Eikon's technology observes proteins in living cells, capturing the influence of cellular factors like chaperones, crowding, and local chemical environments on protein folding and function. The implications of this approach are profound. Instead of inferring folding pathways from final structures or simplified models, scientists can now observe the actual paths proteins take from synthesis to function.
This direct observation has already revealed surprising insights. For example, many proteins fold through multiple pathways rather than a single defined route. Additionally, folding often occurs in discrete steps, with stable intermediates forming along the way5. These observations challenge and refine our theoretical models of protein folding, providing empirical data against which both reductionist models and predictive algorithms can be tested and improved.
Beyond Visualization— Integrating Observation with Intervention
What makes Eikon's approach particularly powerful for drug discovery is the integration of observation with intervention. By simultaneously tracking protein dynamics and testing how small molecules affect these dynamics, researchers can identify compounds that modulate protein function in specific ways.
This approach has already yielded promising results for previously "undruggable" targets. For example, by focusing on dynamic behavior rather than static structure, Eikon has identified compounds that affect protein function by altering their movement patterns, binding kinetics, or conformational transitions, opening new avenues for therapeutic development.
The visualization approach also offers unique insights into disease mechanisms. Many diseases involve abnormal protein dynamics rather than simply abnormal structures. In neurodegenerative diseases like Alzheimer's and Parkinson's disease the process of protein aggregation—how normally soluble proteins form insoluble clusters—is critical to disease progression. Direct visualization of these aggregation processes provides new opportunities to intervene before irreversible damage occurs.
The Future— Integration and Application
The next great leap in structural biology will be defined by the integration of reductionist models, predictive algorithms, and direct visualization techniques — each of which provides unique and complementary insights. Reductionist models teach us about fundamental principles and help us generate hypotheses while predictive algorithms provide rapid access to potential protein structures, guiding experimental design. Finally, next-generation visualization techniques reveal actual protein dynamics, validating, challenging, and stress-testing our models and predictions.
The integration of these approaches holds the potential to transform multiple fields, from protein design to drug discovery and personalized medicine. For example, by understanding how genetic variants affect not just protein structure, but protein folding processes, we may be able to better predict which variants will cause disease and design targeted interventions to cure them. Cystic fibrosis, in particular, comes to mind as a disease that can be treated with this approach. Many cystic fibrosis-associated mutations affect the folding efficiency of the CFTR protein rather than its final structure. Visualizing these effects could lead to personalized treatments that address the specific folding defects in individual patients.
Additionally, integrating structural prediction and dynamic visualization creates new opportunities for drug discovery. Structure prediction can identify potential binding sites and generate candidate molecules, while visualization can reveal how these molecules affect protein dynamics in living cells. This combined approach promises to increase the success rate of drug development by ensuring that candidate drugs have the desired effects on protein behavior, not just protein structure.
Finally, for designed proteins to function as intended, they must not only achieve the correct final structure but fold efficiently and behave appropriately in cellular contexts. By integrating predictive design with dynamic visualization, protein engineers can create proteins that not only look right but work right, expanding the applications of designed proteins in medicine, materials science, and biotechnology.
A New Chapter in Protein Science
The evolution from reductionist models to predictive algorithms to direct visualization represents a fundamental shift in how we approach protein structure and function. Rather than seeing proteins as static objects to be modeled or predicted, we now recognize them as dynamic entities whose behavior emerges from complex interactions over time.
This shift parallels broader changes in biological science, moving from reductionism to systems thinking, from static models to dynamic processes, and from inference to direct observation. As we continue this journey, we can expect not only deeper scientific insights but also practical applications that transform medicine, biotechnology, and our understanding of life itself.
Did you enjoy this piece? If so, you may also want to check out other articles in Decoding Biology’s Bioinformatics & Biotechnology collection.
In the context of protein length, "mer" indicates the number of amino acids or residues in a protein. For example, a 100-mer peptide would be a peptide with 100 amino acids. The term "k-mer" can also be used, where "k" represents the number of residues/amino acids
Homopolymers are molecular chains composed of a single subunits whereas heteropolymers are made up of varying subunits. All proteins are heteropolymers because they are made of different amino acids. An example homopolymer, on the other hand, would be starch.
This transition requires a huge mental frameshift — it suggests that understanding in biology doesn’t only come from reducing systems to components, but also from building models that can anticipate or predict how biological systems will manifest or respond to different conditions.
If anyone from Eikon therapeutics is reading this please sponsor me. As a computational biologist named Evan Peikon, your brand is literally in my name. You won’t get another opportunity like this.
One of the most interesting facts I learned when writing this piece is that some proteins undergo repeated cycles of folding and unfolding on the way to their native state.